
机器学习在化合物属性预测中的应用*
2024-03-01 10:44王紫维
环境化学 2024年1期
王紫维 韩 民 金 彪
(1.中国科学院广州地球化学研究所,有机地球化学国家重点实验室,广州,510640;2.中国科学院深地科学卓越创新中心,广州,510640;3.中国科学院大学,北京,100049)
化合物的属性预测在药物研发、材料设计、毒理学研究等领域发挥了重要的作用,与人类生活息息相关[1−2].化合物属性预测的相关研究可追溯到药物合成的早期研究,当时主要是化学家通过重复实验,进行测试和验证并获取各类化学信息,合成目标分子[3].由于重复实验耗时长、成本高,科学家基于构效关系(SAR)发展出了定量-构效关系,为化合物结构与其性质之间建立了数学关系框架.1962年,Hansch等首次实践了定量-构效关系(QSAR),成为该领域具有里程碑意义的事件,也是化合物属性预测研究迈入新阶段的标志[4].随后,Hansch 在1964年提出了Hansch 方程,这个发现为QSAR 模型运行提供了一种新方法.但传统QSAR 模型一般使用一些常见的分子描述符来预测化合物属性,然而化合物结构多样,少量的分子描述符很难全面地描述化合物的结构信息,这使得模型很难精准预测化合物性质.同时,随着研究数据集增大、描述符增多,传统的方法难以拟合化学结构与性质之间的复杂关系.因此,需要比传统统计工具更先进、更强大的计算和数据分析方法.
机器学习(特别是深度学习),由于其强大的计算和数据分析能力,已被用于解决以上QSAR 研究中的问题.例如,研究人员通过机器学习或深度学习方法将三维甚至更高维分子结构与其属性联系起来,弥补了传统的化合物属性预测方法的不足之处,大力推动了化合物属性研究的发展[5−6].
近年来,机器学习在化合物属性的预测研究上表现出不俗的潜力,因此这方面的研究也逐年增多.比如在理化性质方面,在机器学习的帮助下,预测分子的原子化能、振动频率、溶剂化自由能、计算键离能等,成本更低,结果准确可靠,计算速度更快[7−11];在生物活性方面,建模方面逐步引入了神经网络算法、分子图等,所构建模型性能更优异,结果可靠[12−14];在毒性方面,根据机器学习建立的模型可以非常有效地识别有毒分子和预测特定毒性,可筛选确认之前未曾识别出的危险化学品[15−17].本文主要介绍机器学习在化合物属性预测方面的应用过程及相应的模块内容,并结合应用实例总结和展望机器学习在该应用方面现存的问题和机遇.
1 机器学习预测化合物属性的流程(The process of machine learning on compound property prediction)
在实际应用中,用机器学习预测化合物属性的整体过程如下所述,见图1.
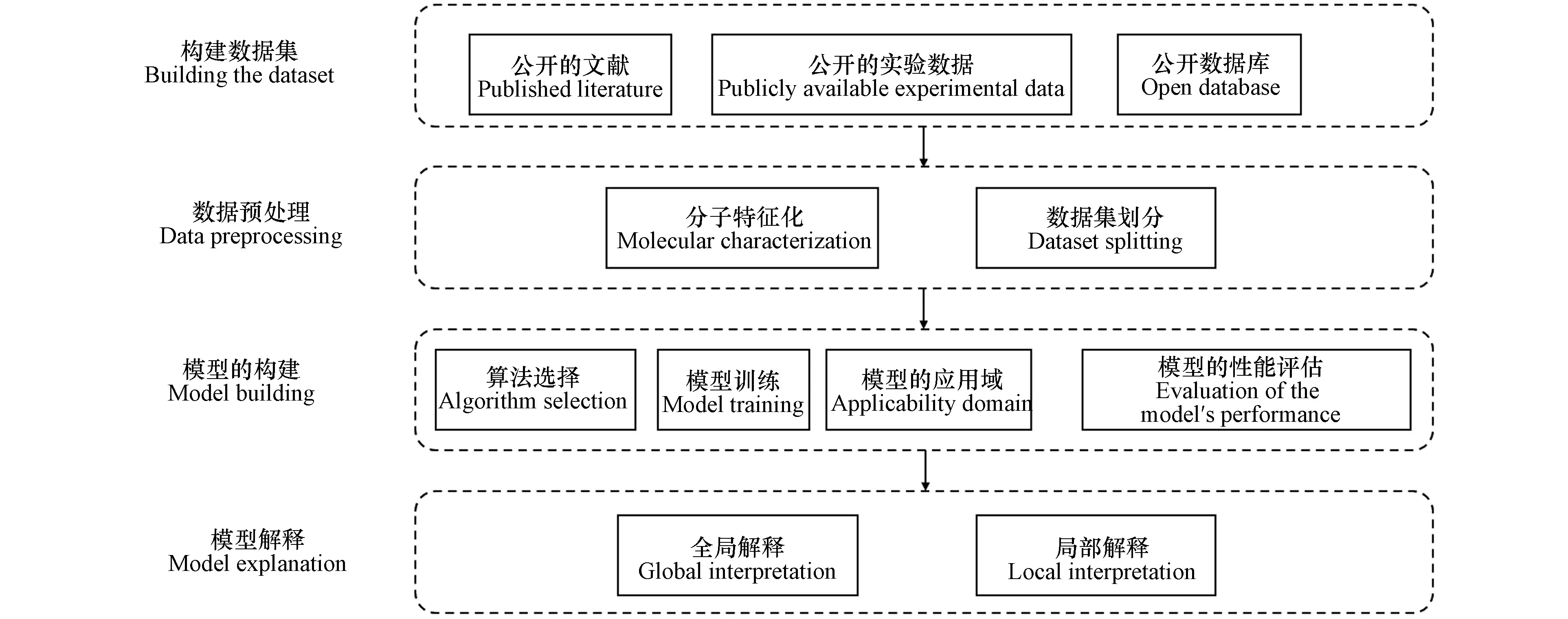
图1 机器学习进行化合物属性预测的流程Fig.1 Process for compound property prediction based on machine learning
(1)构建数据集:数据集一般来源于公开的数据库、实验记录数据、研究收集的文献;
(2)数据预处理;主要包括分子特征化和数据集划分;
(3)模型构建:主要包括模型训练、算法选择、模型的应用域、模型评估这四方面内容;
(4)模型解释:解释机器学习模型的预测机制.
1.1 构建数据集
构建数据集是构建模型进行化合物属性预测之前的必要准备,模型的工作主要基于数据运行,数据集的质量影响了模型预测结果的可靠性以及准确性.目前众多研究工作一般通过以下几种途径进行数据集的构建:(1)根据实验所得数据构建数据集;(2)从公开的数据库中下载研究所需要的数据,并整理成数据集;(3)提取他人发表在期刊、专利中的数据,整理成数据集;(4)前述3 种方式的组合形式.
实验室所记录的数据比较全面,是构建数据集的来源之一.实验所得数据一般分为纸质记录数据和电子记录数据,其中,电子记录数据可用于数据集构建和数据挖掘,但由于实验数据为实验者所有,大多用于进行数据存档和知识产权维权,难以获得全面的数据来进行数据挖掘.同时,他人发表在期刊、专利中的数据相对于实验所记录的数据大多数都经过文章作者精心筛选,没有展示相关实验失败的数据.失败数据的缺乏可能会造成信息缺失,从而导致构建的模型不能进行准确地预测.
构建数据集最常用的方法是从公开的数据库获取研究要用的数据.经过多年的发展,目前也有许多可免费获取化学数据的公开数据库,部分较常见的公开数据库可见表1.

表1 常见的公开数据库Table 1 Common public databases
1.2 数据预处理
1.2.1 分子特征化
分子特征化是把化合物的化学结构编码成机器学习算法能识别的模式.不同的分子特征化方式提取的分子信息有所差异,直接影响模型的预测效果,因此是化合物属性预测的重要部分.常见的分子特征化方法有分子描述符、分子图、分子线性表示、分子图像[31],以及三维分子表面点云[32].
(1)分子描述符
分子描述符与分子结构的关系密切,可以有效地表示相应的化学信息[33].分子描述符按照复杂程度,可分为零维、一维、二维、三维等(见图2)[34].零维描述符是最简单的分子描述符,其信息含量低,可表示原子数、原子性质总和、分子量等;一维描述符表示一些官能团、分子片段、取代基等信息,如分子量、摩尔折射率、辛醇/水分配系数的对数等;二维描述符可描述从二维分子表示计算得到的性质;三维描述符信息含量很高,可描述原子的性质、连通性以及分子的空间构型,可用于确定化合物的活性构象等问题;四维描述符可以定量识别和描述分子与受体活性位点之间的相互作用[34].

图2 同分子的不同分子表示的示例[34]Fig.2 An example of different molecular representations of the same molecule[34]
分子描述符按照定量和定性分类,可分为定量分子描述符和定性分子描述符.定量分子描述符有分子场描述符、分子形状描述符、物理化学描述符、基于组成信息的描述符等[35].定性分子描述符一般指分子指纹,分子指纹又称二元指纹,采用二进制编码相关的化学信息,指纹所具有的化学信息内容一般为化学图中的原子、键类型和距离等,是化学结构的表示,常被用于分子相似性/多样性问题[34,36].
常见的分子指纹可主要分为基于子结构的指纹、基于拓扑或路径的指纹和圆形指纹、药效团指纹等[37].基于子结构的指纹主要有MACCS 指纹[38]、PubChem 指纹、BCI 指纹、TGD 和TGT 指纹等.基于拓扑或路径的指纹主要有Daylight 指纹(Daylight fingerprint)和Tree 指纹(Tree fingerprint).圆形指纹主要有扩展连通性指纹(ECFP/Morgan Fingerprint)[39]、FCFP(Functional-Class Fingerprints)、Molprint2D[40].
常用于计算分子指纹的软件或工具包有alvaDesc[41]、RDkit、Open Babel[42]、CDK[43]、ChemFP、OEChem TK、Molecular Operating Environment(MOE)、JChem from ChemAxon、Pipeline Pilot from Accelrys 等.
(2)分子图
分子图是指化合物用图进行表示,是化合物的拓扑表示.在分子图中,原子用节点表示,分子键用边表示,示例可见图3.分子图降低了分子结构表示的复杂性,可以捕捉到分子中原子核与电子间的关键的相互作用.此外,图神经网络(GNN)模型从分子图进行学习表示可以得到很好的处理效果,减少了相应的特征工程的工作,能进行更好的分子性质预测,如Attentive FP[44]、D-MPNN[8].
(3)分子线性表示
分子线性表示最常用的有两种:简化分子线性输入规范(Simplified molecular input line entry system,SMILES)和国际化合物标识符(International Chemical Identifier,InChI).SMILES 是一种利用ASCII 编码表示分子结构的线性符号,是化学家为了进行化学方面的机器计算而设计的化学符号语言,是根据相应的规则对化学结构简化的二维价键图[45].SMILES 既可以与化学数据库使用,又可以节省存储空间,为化学数据的输入提供了一种更简便的方式.化合物的“SMILES”字符串可通过一些软件或程序获得,如ChemDraw、OpenBabel、CIRpy[46](https://github.com/mcs07/CIRpy)等,同时也可以通过网站获得化合物的“SMILES”字符串,如PubChem.“SMILES”字符串除了可以直接作为模型的输入,也可以通过一些软件或程序转换为其他分子特征化形式,再作为模型的输入[6,46−48].通用的SMILES 基于CANGEN算法衍生了规范的SMILES(Canonical SMILES),但其算法具有盈利性质,从而存在无法自由使用的问题.InChI[49]是一个非盈利的、免费的化学标识,在描述分子方面具有严格的唯一性,在层状设计时考虑了分子结构,容易获得和生成,可以由InChI 软件或者利用通用的化学绘图软件生成.因此,InChI 也被许多化学数据库使用.
(4)分子图像
分子图像是将分子结构或坐标映射到图像上后,作为模型的输入数据用于模型训练,从而进行分子性质预测[50].比如,可以通过OpenBabel、Pybel 和RDKit 等化学信息软件将SMILES 解码为对应的分子二维结构,再将其生成的坐标映射到网格上,形成分子图像,示例可见图4.对于所生成的图像可再进行一个“灰色编码”或者更为复杂的“颜色编码”,表示出原子/键属性,再用于卷积神经网络(CNN)算法进行训练[50−51].
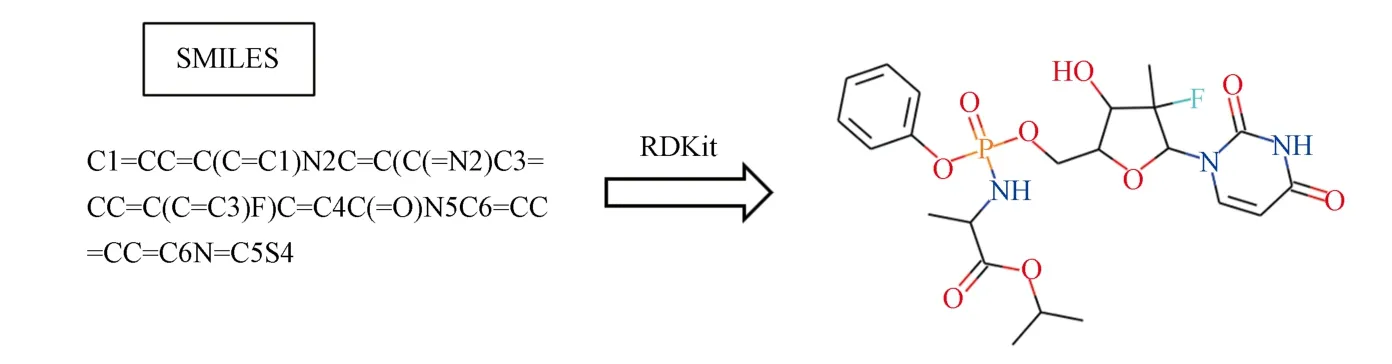
图4 RDkit 将化合物SMILES 转换为分子图像的示例Fig.4 An example of RDkit transforming SMILES into the molecular image
1.2.2 数据集划分
整理好研究所需的数据集之后,应及时对数据进行划分,数据划分对于所建机器学习模型的泛化能力有一定影响.一般,数据集按比例随机划分为3 部分:训练集、测试集、验证集,其中,训练集一般用于模型的训练,测试集用于模型的性能评估,验证集用于超参数的优化[52−53].但是,研究过程中数据集划分的具体的分配比例应按照研究需要进行选择,比如,有研究按8:1:1 的比例划分成训练集、验证集和测试集[47];也有研究先将数据集按4:1 的比例随机分成两部分:训练集和测试集,随后在训练过程中随机抽取10%的训练集数据作为验证集数据[52].
当机器学习应用于分类问题时,可能会碰到数据集中各类别样本数量分布不均衡的问题,即数据集中某一类别的样本数量远远高于另一类.不平衡数据会影响模型的性能,因此,在数据集划分后需对训练集数据进行不平衡处理.目前进行不平衡数据处理的方法主要有数据重构和分类模型改进.
数据重构策略可分为特征选择和重采样技术[54].特征选择方法主要分成3 类:过滤式、包裹式和嵌入式.重采样技术是通过调整多数类和少数类的分布,削弱数据集不平衡的程度,主要包括欠采样、过采样、混合采样.欠采样通过减少多数类中的样本数量,以平衡多数类和少数类;过采样通过增加少数类中的样本数量,以均衡数据集;混合采样通过将过采样和欠采样组合在一起,提高分类性能[52].
分类模型改进策略主要从分类算法和分类思想对不平衡数据集进行优化和改进.分类算法主要有K 最近邻、支持向量机、决策树、朴素贝叶斯、神经网络算法;分类思想主要有代价敏感学习、集成学习、单类学习、主动学习[54].
1.3 模型的构建
1.3.1 算法选择
模型训练是指通过训练集数据进行拟合模型、学习模型参数的过程.在这个过程,需要选择合适的算法进行训练.算法是机器学习的核心内容,没有算法,机器学习模型将无法运行.目前,机器学习的算法按照是否有人类监督训练,可分为监督式学习、无监督式学习、半监督式学习、强化学习这4 个主要类型.监督式学习可以处理有标记的训练数据,其算法主要有K-近邻算法、线性回归、逻辑回归、支持向量机(SVM)[55]、神经网络、决策树[56]和随机森林(RF)[57].无监督式学习使用的数据是没有标记的,其算法主要可分为聚类算法、可视化和降维算法、关联规则学习算法.聚类算法主要有k-平均算法、分层聚类分析、最大期望算法等;可视化和降维算法主要有主成分分析(PCA)、核主成分分析(Kernel PCA)、局部线性嵌入(LLE)、t-分布随机近临嵌入(t-SNE);关联规则学习算法主要有Apriori、Eclat.半监督式学习可以处理部分标记(大量未标记和少量标记)的数据,其主要为监督式学习算法与无监督式学习算法的结合.强化学习[58]是学习到行动的一种映射,通过不断试错,寻找到能够最大化预期的路径,并对能最大化预期的行动进行奖励,主要涉及到的策略是试错搜索和延迟奖励,这两种策略是强化学习的最明显最重要的特征.在化合物属性预测中,常用的是监督学习模式[59]、半监督式学习;常用的算法有随机森林[57]、支持向量机[55]、朴素贝叶斯、神经网络、梯度提升决策树(GBDT)、极限梯度提升算法(XGBoost)、线性回归、决策树[56]、逻辑回归等算法.
1.3.2 模型应用域
在化学信息研究中,为了更高效地进行化合物属性的预测,通常将机器学习方法应用于定量构效关系中,其中涉及到的模型应用域(AD)一般指化学空间中由描述符和模型响应定义的一个理论域,其任务是定义一个模型可以被使用的边界,并提供可靠的预测[60−61].当要预测的分子在所定义的AD 范围内时,使用该模型进行预测才可靠,否则该模型不适用.
对于模型应用域的表征,已有学者在这个方面进行过总结,不同的学者考虑的角度不同,进行的归类方式和描述称呼各有不同.Kar 等[61]根据不同假设将AD 的定义方法分为6 大类:描述符空间中基于范围的方法、基于距离的方法、几何方法、概率密度分布、响应变量的范围、其他方法,而王中钰等[62]根据AD 的概念将其分为描述符域、结构域、机理域3 个大类,再从这3 大类中对AD 的表征方法进行细分.此外,也有一些学者提出或开发了新的应用域表征方法或应用域算法,如Wang 等人[63]基于指纹特异性相似性阈值,开发了新的AD 表征方法—ADfingerprint,并证明其性能优于一些传统的AD 表征方法;Berenger 等[64]对于创建的应用域较为复杂并难以理解的现状,提出了基于距离的Boolean 应用域算法(DBBAD).虽然AD 表征方法的描述各有异同,但最常用的几种表征方法一般为欧式距离、Tanimoto指数、杠杆方法、马氏距离、核密度估计(KDE)、基于范围的超矩形等方法.连续数据的研究,一般选用欧式距离定义模型应用域[65];而针对二进制数据或者想要进行分子相似度的比较的研究,一般选用Tanimoto 指数定义模型应用域[47−48].
1.3.3 模型性能评估
模型的性能评估是对所构建的模型的泛化能力进行评估,有助于判定模型的工作性能和开发适合研究数据的最佳模型,主要包括了性能度量、评估方法、过拟合或欠拟合、超参数调优、泛化能力这几个方面的内容.模型泛化能力是指所构建的模型经过在训练集数据的训练之后,在新数据上的适应能力.过拟合和欠拟合都是模型泛化能力不好的行为表示.过拟合指模型过度学习训练集数据,使得模型过为复杂,不能在除训练集外的数据集上得到好表现;欠拟合指模型过为简单,学习能力差,无法学到数据的内在特点,无法判定其潜在的趋势[66].
模型性能的评估方法常见的有留出法(hold-out)、交叉验证法(cross validation)、自助法(bootstrapping).在模型评估的实际情况中,一般先用评估方法对模型进行数据集划分,再在测试集上用评估指标对模型性能进行评估.比如,Korkmaz[52]在研究中先用留出法将数据集划分为80%的训练集和20%的测试集,之后用计算了几个性能指标对模型进行了性能评估.
在化合物属性预测方面的应用,主要可将机器学习任务分为回归问题和分类问题两方面.在性能度量涉及到的性能指标方面,回归问题和分类问题所用到的性能评价指标并不完全一致.回归问题常用到的评价指标有均方根误差(root mean squared error,RMSE)、平均绝对误差(mean absolute error,MAE)、均方误差(mean square error,MSE)、均方根对数误差(root mean squared logarithmic error,RMSLE)、决定系数(coefficient of determination,R2)和预测平方相关系数(predictive squared correlation coefficient,Q2)等.分类问题常用到的评价指标有准确率(accuracy,acc)、错误率(error)、精确率(precision rate,p)、召回率(recall rate,r)、F1 分数(F1-score)、ROC 曲线(receiver operating characteristic)、AUC(area under curve)等.分类问题的数据集并不一定平衡,在大多应用情况下都会出现数据分布不均导致数据不平衡的现象,这种情况下,首先要在训练集上进行数据不平衡处理,之后再用测试集对其进行评估.针对不平衡数据集,准确率往往无法作为主要的判断指标,因此一般可采用前文所提到的精确率、召回率、F1 分数以及均衡准确率(balanced accuracy)和G-mean[54].除了上述的指标外,还有一些其他的指标,如鲁棒性、PRC(精确-召回曲线)等.在实际的应用情况中,指标的选择应根据数据的情况和研究需要来进行选择.
1.4 模型解释
模型解释是对模型的预测机制进行解析的过程,有利于研究者做出更好的决策,并建立起对模型的理解和信任[67−68].模型根据解释的难易程度,可以分为“白盒”模型和“黑盒”模型.“白盒”模型又可称为可解释性模型,创建其模型的算法透明度低,解释简单,更易被人们理解.可解释性模型一般指由线性回归、逻辑回归、其他线性扩展、决策树等算法构建的模型.建立“黑盒”模型后再进行解释这一行为也可称为事后可解释性,进行事后可解释的方法主要可以分为两大类:全局解释和局部解释,全局解释是对模型整体进行解释,而局部解释是对单个预测进行解释[69].
全局解释的方法主要有部分依赖图(partial dependence plot,PDP)、累积局部效应(accumulated local effects plot,ALE)、规则提取[70]、模型蒸馏[71]、稀疏集团套索(sparse group lasso,SGL)[72]、全局Shapley值等.局部解释的方法主要有个体条件期望(individual conditional expectation,ICE)[73]、敏感性分析、局部可解释的模型无关阐释(local interpretable model-agnostic explanations,LIME)[67]、Anchor[74]、基于局部规则的黑盒模型的分层相关性传播(LRP)[75]、类激活映射(class activation mapping,CAM)、梯度加权类激活映射(Grad-CAM)[76]、SHAP(shapley additive exPlanations)[77]等.在化合物属性预测方面,比较常用的解释方法有PDP、ALE、ICE、Grad-CAM、Shapley Value、SHAP 等.如Zhong 等[6]利用Grad-CAM来解释构建的CNN 模型通过选择分子图像的哪些特征来进行预测.Sanches-Neto 等[46]在预测水中有机污染物自由基氧化过程的反应速率常数的研究中,利用SHAP 方法解释了反应过程中相关的结构分子特征,将氧原子所做的贡献从氧原子与碳原子的比例(#O:C)的贡献区分出来.
2 机器学习在化合物属性预测中的应用进展(The application progress of machine learning on compound property prediction)
2.1 理化性质预测
机器学习中的神经网络算法可被用于量子化学性质预测.比如,2017年,由Gilmer 等[7]提出来的应用于分子图的监督学习框架——消息传递神经网络(message passing neural networks,MPNNs),更易理解图的结构数据与模型之间的关系.他们基于MPNNs 进行建模,采用QM9 数据集的数据,对分子的原子化能、振动频率、最高占据分子轨道(HOMO)、最低未占据分子轨道(LUMO)、偶极矩等性质进行了预测,结果表明利用机器学习进行分子性质预测的成本比密度泛函理论(DFT)计算低且计算速度更快,计算样本误差比DFT 小,在大型图中应用良好.之后,有学者在MPNNs 的基础上进行改动,提出了知识嵌入消息传递神经网络(KEMPNN)[78].KEMPNN 在MPNN 中的消息传递阶段添加了知识注意机制作为一项加权项,采用两个数据集共同训练MPNN,并在ESOL,FreeSolv,Lipophilicity 以及聚合物性能数据集上进行了测评,与MPNN 进行了对比.结果表明,KEMPNN 比MPNN 的模型的预测精度更高,并且发现了KEMPNN 在小数据集上的预测效果可与基于描述符的方法相当甚至更好.
溶剂化自由能与许多物理化学性质密切相关,在药物发现方面有重要的影响,但溶剂化自由能的实验数据较少,且实验成本昂贵.尽管已经有一些相应的溶剂模型可预测溶剂化自由能,使得费用成本有所降低,但其准确性较低.相比之下,机器学习在溶剂化自由能预测方面更具优势,既不会产生昂贵的费用,又保证了较高的溶剂化自由能预测准确率[10,79].如,Yang 等[8]在MPNN 的基础上构建了一个基于定向键的消息传递方式,并结合分子水平特征和分子式构建了新的模型D-MPNN,在FreeSolv 数据集上表现出比其他基准模型更好的性能;Weinreich 等[9]提出了一个以核岭回归(KRR)算法作为监督机器方法的自由能机器学习模型(FML),并在FreeSolv 数据集和QM9 数据集上进行了溶剂化自由能预测,模型误差与最好的物理预测方法相当,但计算成本更低,并且可在较小数据集上达到溶剂化的实验不确定度.需要指出的是,机器学习在溶剂化自由能预测方面存在数据稀缺的问题,深度学习的模型在小数据集上容易过拟合,性能差.鉴于此,Vermeire 等[80]基于D-MPNN 构建了一个模型,通过引入一种结合量子化学和实验数据的迁移学习方法使模型在溶剂化自由能预测方面的性能得到了显著提升;Zhang 等[10]提出一个基于GNN 和3D 原子特征的深度学习(DL)模型构架,GNN 以主领域聚合PNAConv 作为编码器,并将其与迁移学习策略相结合,进行模型微调后在FreeSolv 数据集进行溶剂化自由能预测并得到了目前最好的性能,RMSE 为0.719 kcal·mol−1,MAE 为0.417 kcal·mol−1,显著提高了GNN 模型在溶剂化自由能预测方面的学习能力,为处理小型实验数据集提供了思考方向.
此外,机器学习在预测化合物的其他性质方面也有不错的表现,以全氟化合物(PFASs)理化性质预测为例.在全氟化合物(PFASs)理化性质预测方面,Raza 等[11]在2019年提出了第一个利用机器学习来预测各种PFAS 结构中的C—F 键解离能的应用.这个应用高效可靠准确,训练数据时间短,预测C—F 键解离能的时间不超过1 s,偏差小于0.70 kcal·mol−1,不需量子力学计算,计算成本更低,有助于PFAS 和高效处理与去除.之后,有学者[81]于2021年构建了一个数据库框架,所构建PFAS-Map 可以预测未测定的PFAS 化学品的基本物理性质,可视化PFAS 活性/性质关系的实验数据趋势,发现隐藏的结构 -毒性关系.
2.2 生物活性预测
机器学习在上世纪就开始用于进行生物活性预测.在20 世纪90年代,神经网络算法广泛应用于定量结构-活性关系,但由于其算法的局限性,在2000年早期被SVM 和RF 取代.近些年,神经网络算法逐步改进,引起了人们的关注,发现改进后的神经网络算法在生物活性预测方面颇具优势.2015年,Ma 和Dahl 等[82]采用“原子对”描述符和“供体-受体对”描述符的并集作为描述符来训练模型,并将深度神经网络(DNN)的性能评估参数R2与RF 模型在15 个数据集(Merck 公司内部的数据集)上进行比较,结果表明DNN 在大多数情况下预测性能都优于RF 模型,在计算时间和成本方面甚至比RF 更有优势,可作为一种实用的QSAR 方法.但需要指出的是,该项研究也存在局限性,无法阐明分子间未完成的潜在相互作用.针对这些缺点,Wallach 等[12]建立了第一个基于结构的深度卷积神经网络—AtomNet,可应用于小分子生物活性预测.他们将AtomNet 与DNN 技术进行对比,发现AtomNet 可为目标预测出新的活性分子,所构建的模型能发现任意的分子特征,可描述配体和目标之间的相互作用;同时,在3 个基准上做了应用,结果表明AtomNet 表现出色,在DUDE 基准测试中有一半的目标的AUC 为0.9,远超以前的对接方法.
此外,2019年,Cheng 和Ng[13]在前人的基础上建立了ML-QSAR 模型预测全氟化合物(PFASs)的生物活性,引入了基于图的模型,预测了OECD 名单中未经测试的PFASs 的生物活性.在整个过程中,基于自行收集整理的PFASs 数据库训练和评估了5 种机器学习模型,采用了ECFP、图卷积、weave 特征3 种方法进行分子特征化,网格搜索和贝叶斯优化技术进行超参数调优,基于距离的方法确定QSAR 模型的AD 值,结果表明,多任务神经网络模型和基于图的图卷积模型性能优异,但构建的模型不能提供有关效应强度或剂量反应的信息,有进一步发展的空间.此外,不同于常用于化合物活性预测的结构-活性关系(SAR)模型,Bertoni 等[14]于2021年构建了一个深度神经网络的集合—SigAR(signature–activity relationship)模型预测分子的生物活性,让机器学习从化合物的CC signatures(基于一个小分子生物活性特征集合开发的分子表征方法)中学习活性特征,并用MoleculeNet 中的9 个数据集评估了SigAR.其结果表明,相较于基于化学描述符的方法,SigAR 的性能更好.
2.3 毒性预测
对化合物的毒性进行预测,是药物研发的一部分,对于药物研发的成本和成功率有重要影响.同时,化合物毒性预测也是化学品风险评估的一部分内容,但基于动物实验的毒性预测,时间周期长,成本开支大.此外,人工合成化合物的种类在逐渐增多,在日常生活中随处可见,识别危险化学品的潜在毒性是有必要的,对化合物进行毒性预测的需求在持续增长.机器学习应用于化合物的毒性预测具有降低成本和加快研究速度的特点,因此,机器学习在化合物毒性预测方面的研究一直以来都是热点研究领域,相关的研究也比较多.
2008年,美国的EPA、NIH 和FDA 开展了Tox21 计划,这个计划汇总了许多化合物的毒性数据,推动了机器学习在预测化学品的潜在毒性和评估化学品风险的进程.2016年,Mary 等[83]开发了适用于毒性预测的集成模型—DeepTox,并将其运用于Tox21 挑战赛上.他们采用了化合物的大量的静态特征(如,MACCS 指纹、PubChem 子结构指纹等)和动态特征(如,ECFP 指纹、径向2D 指纹等)作为机器学习的输入,并对DeepTox 中的每个机器学习算法模型进行了性能评估,比较了各算法的AUC值,结果表明DNN 优于SVM、RF、弹性网(ElNet).同时,由DNN 主导的DeepTox 应用于预测化合物毒性,取得了Tox21 大挑战的冠军.2019年,Pu 等[84]基于机器学习技术开发了一个新的程序—eToxPred,可以直接从分子指纹预测小型化合物的毒性.eToxPred 采用额外树(Extra Trees,ET)算法作为毒性预测的默认分类器,并在不同的数据集上与线性判别分析(LDA),多层感知器(MLP),随机森林(RF)算法进行了性能对比.结果表明,使用分子指纹作为输入,基于ET 的分类器性能普遍高于LDA 和MLP,仅在一个组合数据集上略低于RF,可以非常有效地识别有毒分子和预测特定毒性.
在化合物毒性评估方面,常用结构警报(structural alerts,SAs)作为识别危险化学品的潜在毒性的方法,但SAs 的准确性有限,有时在无毒化合物中也会发现SAs[85].Mukherjee 等[15]引入了一个新概念——“关键结构图案”(critical structural motif,CSM),CSM 包含了SAs 的特异性.同时,他们用SMILES 字符串作为模型输入,开发了一个基于卷积神经网络(CNN)的多输出分类的深度学习模型--VisualTox,并在不同的化学数据上进行了训练,通过识别 CSM 来预测内分泌干扰物质(ECD)的毒性,提供了一种理解化学毒性来源的新方法.
此外,持久性有机污染物(POPs)和持久性、生物累积性和毒性物质(PBT)对生态环境和人类健康都有重大影响,PBT/POP 类化学品也备受人们的关注.Sun 等[16]于2020年采用基于2424 个分子描述的二维表示矩阵(MDRM)作为模型输入,开发了一个深度卷积神经网络(DCNN)模型来筛选化学品库中潜在的PBT/POP 类物质,并采用k折交叉验证法和专家经验判断方法对模型性能进行评价,得到模型的预测精度可达90.4%.但需要指出的是,DCNN 模型是一个“黑盒”模型,基本不可得到有效的解释.最近,Wang 等[17]利用一个包含14994 种PBT 和non-PBT 物质的化学数据库,基于图注意力网络(graph attention networks,GATs)架构,构建了可筛选PBT 化学品并具有可解释性的GAT 模型.GATs 是一种较先进的GNN,为分子图的每个节点引入了注意权重参数(PAW),可反映节点对预测端点的贡献,关注与目标任务相关的重要局部结构,具有模型可解释性.他们在AD 表征方面,提出并采用了一种新的方法—ADFP-AC,使GAT 模型更加可靠;在模型性能方面,将具有ADFP-AC表征的GAT 模型与DCNN 模型、传统的机器学习方法(如随机森林、支持向量机)和根据不同分子特征化方法建立的QSAR 模型进行性能对比,发现GAT 模型的性能最佳.在建立好GAT 模型之后,他们还将其应用在中国现有化学物质清单(IECSC)上,从中确定了8 类之前未确认的化合物类别为PBT 化学品.
3 机器学习在化合物属性预测中的挑战(The challenges of machine learning on compound property prediction)
3.1 数据集
目前,在构建数据集的过程中,研究者往往面临以下3 个问题,包括数据量不足、数据质量不高以及数据不平衡.针对数据量不足问题,虽然前文介绍了一些相关的公开数据库,但这些数据库对于研究人员来说,数量还是较少,而且数据不够全面,很多重要的化学信息被收集在商业数据库中或其他难以获取的数据库中.此外,虽然公开的一些大型化学数据库数据多,规模大,但是拥有的标签数据并不多.这种情况限制了监督学习在化合物属性预测方面进行更深入的研究[51].这些都使得研究人员无法得到足够的数据,利用机器学习在化合物属性层面进行一个更好的突破.面临的问题之二是数据的质量不高.有些数据来自于实验记录,虽然实验记录数据能得到更多、更为全面的数据,但公开的实验数据如何保证质量,也是值得思考的问题.面临的问题之三是数据不平衡问题.虽然机器学习有许多经典的分类算法,如朴素贝叶斯、KNN、基于神经网络的分类算法等,这些算法尽可能地保留了原数据所有的信息,但是由于这些算法的假设都是基于平衡的样本数据,所以当数据有少数类和多数类的情况出现时,这些算法皆会更倾向于多数类数据[86].对此,许多学者提出了一些数据不平衡处理方法,如过采样、欠采样、混合采样和特征选择等,这些处理方法在一定程度上能够缓解不平衡问题,但都存在不足.比如,欠采样方法虽然简单又效果好,但是容易忽略多数类数据的内在特征信息,影响模型的泛化能力.
3.2 分子特征化
分子特征化方法是化合物属性预测中的重点之一,决定了模型的性能和解释.目前,分子特征化方法能够表征的信息很多,比如,定量分子描述符可以量化Hammett 常数、偶极矩、HOMO 和LUMO 能量等信息,为化合物的性质预测提供了良好的输入信息.但目前还没有可以完整表达原始分子信息的特征化方式[87].
此外,虽然分子指纹种类也颇多,但目前主要还是用二维(2D)分子指纹来做相应的研究,高维度的分子指纹设计较为困难,这导致了现有的分子指纹种类缺少对分子立体结构描述的三维结构信息.对于此类问题,近些年也有学者提出了代数图、代数拓扑、微分几何等分子三维结构信息的表示方法,但是这些方法较为依赖分子结构的可用性[88].分子特征化方法在描述分子的立体化学信息方面还有许多空间可以提升.
3.3 模型的可解释性
模型的可解释性是模型的重要部分,是可信性的前提,如何让模型的工作机制更为透明,获得人们的理解和信任,这是值得讨论和重视的.可解释的机器学习模型没有“黑盒”模型的特征,更易被理解,透明度高.相较于可解释的机器学习模型,具有“黑盒”特征的机器学习模型虽然更难进行直观的解释,但是其性能更高,预测效果更好.因此,如何对“黑盒”模型进行更好的解释,增加模型的可解释性,需要更多的研究来进行探究.同时,现在缺乏明确的模型的可解释性基准,没有严格的方法来评估和比较模型解释方法[89].
4 总结(Conclusion)
机器学习在化合物属性预测方面的应用不断拓展,不仅提高了预测结果的准确性,而且为评估新化学物质的环境风险提供了新方法.其中,深度学习算法更适用于大数据集,而机器学习算法应用在小数据集更具优势.但是,机器学习在化合物属性预测中的应用仍存在未知和挑战,这些亟待解决的问题将是未来研究工作的焦点.机器学习(特别是深度学习)将会与量子力学、毒理学、量子化学、电化学等深度融合,在药物研发、毒理学研究、环境行为预测、材料研发等领域继续发挥重要作用.
猜你喜欢
环球时报(2022-07-13)2022-07-13
环球时报(2022-03-14)2022-03-14
测绘学报(2022年12期)2022-02-13
小哥白尼(趣味科学)(2021年11期)2021-02-28
小天使·一年级语数英综合(2020年10期)2020-12-16
计算机应用与软件(2020年6期)2020-06-16
电影(2018年8期)2018-09-21
数字通信世界(2018年1期)2018-04-18
测绘科学与工程(2017年5期)2017-05-07
自动化学报(2016年8期)2016-04-16
