
KLF4 Protects VSMCs from Damage of Foam Cell-like Phenotypic Transformation by Maintaining Cellular Homeostasis through Supporting PPARγ Signaling Pathway
2024-02-27 10:36:32ZHANGKaiGAOShangLIUDan
中国生物化学与分子生物学报 2024年1期
ZHANG Kai , GAO Shang , LIU Dan
(1)Institute of Chemical Engineering, Shijiazhuang University, Shijiazhuang 050035, China;2)Shijiazhuang Key Laboratory of Target Drug Research and Pharmacodynamic Evaluation, Shijiazhuang 050035, China;3)Department of Biochemistry and Molecular Biology, Hebei Medical University, Shijiazhuang 050017, China)
Abstract Atherosclerosis is popularly recognized as a multifaceted inflammatory disease that associated with varies cells in vascular wall [including endothelial cells and vascular smooth muscle cells (VSMCs)] and circulating as well as resident immunogenic cells (such as monocytes/macrophages). Foam cells formed by cholesterol-overload VSMCs may play an essential role in the progression of atherosclerosis. Krüppel-like factor 4 (KLF4), a key anti-inflammatory transcriptional factor, has been shown a beneficial effect on vascular function, especially in regard of cardiovascular disease. However, it is unclear that whether KLF4 may play a protective role in the damage of cholesterol to VSMCs. The aim of this study was to investigate the role of KLF4 in the process of foam cell-like phenotypic transformation of VSMCs during atherosclerosis progression and its underlying molecular mechanism. The results of atherosclerosis modeling in mice showed that KLF4 disruption accelerated atherosclerosis progression. Intracellular oil red O staining and cholesterol content assay confirmed that loss of KLF4 promoted cholesterol accumulation in VSMCs. The results of qRT-PCR and Western blot confirmed that KLF4 deletion promoted cholesterol uptake, synthesis, pro-inflammatory factor secretion, macrophage adhesion and expression of macrophage markers which induced by cholesterol damage, and meanwhile inhibited cholesterol efflux and oxidation-related genes as well as contractive phenotypic markers expression, which finally resulted in VSMCs-derived foam cell formation. Furthermore, rescue expression of KLF4 notably reversed above changes. Additionally, KLF4 blocked the activation of TLR4-MyD88-p65 pathway induced by cholesterol accumulation and upregulated the expression of proteins important for cholesterol efflux. SiRNA-induced selective gene ablation of peroxisome proliferator-activated receptor γ (PPARγ) abolished these effects of KLF4. In conclusion, our results suggest that KLF4 exerts a protective effect on cholesterol stimulation in VSMCs by activating PPARγ pathway, inhibiting cholesterol uptake and synthesis, accelerating cholesterol efflux, and meanwhile preventing pro-inflammatory cytokines secretion, macrophage adhesion, and macrophage markers expression.
Key words Krüppel-like factor 4 (KLF4); peroxisome proliferator-activated receptor γ (PPARγ); atherosclerosis; vascular smooth muscle cells (VSMCs); foam cell-like phenotypic transformation
Cardiovascular disease (CVD) has been recognized as a prevailing risk of human health around the world and atherosclerosis is known as the basis for the pathogenesis of various CVDs[1,2]. Thrombosis and obstruction of coronary artery caused by atherosclerotic lumen-narrowing and plaque rupture are the main reasons of sudden and unpredictable onset of acute coronary syndrome[3,4].
Vascular smooth muscle cells (VSMCs) are the main components in the media of artery wall. In the condition of diverse environmental factors stimulation, VSMCs exhibit an ability of phenotypic transition from contraction to proliferation which presenting reduced contractile biomarkers while increased proliferative, secretory as well as migration abilities[5,6]. Evidence has demonstrated that phenotypic transformation of VSMCs may play a critical role in the pathogenesis of various cardiovascular diseases (CVDs), ranging from atherosclerosis and post-injury restenosis to vascular calcification and aneurysms, etc[7-9]. Recently, accumulated evidence has indicated that VSMCs are a significant origin of foam cells and the relative contribution of VSMCs to total foam cells has been determined in human atherosclerosis and in the model of mice[10-12]. The vital role of VSMCs in the progression of atherosclerosis has hitherto been underrated. However, the underlying mechanism of foam cell-like phenotypic transformation of VSMCs remains to be unclear.
KLF4 is one of the Yamanaka factors which is necessary for embryonic cell pluripotency[13-15]and plays a foremost transcriptional factor role in governing anti-inflammatory phenotype of both endothelial cells (ECs) and monocytes/macrophages. Physiological stimulus (e.g., atheroprotective flow) and cardiovascular medications (e.g., statins) exert their favorable effects in part through the induction of KLF4 in ECs and monocytes[16-18]. Under pulsatile shear stress, KLF4 transcriptionally up-regulates many atheroprotective genes such as endothelial nitric oxide synthase (eNOS), thrombomodulin, and inositol 1,4,5-trisphosphate receptor 3 (ITPR3)[19-21]. Furthermore, induction of KLF4 favors the transition from M1 to M2 phenotype in the macrophage, which transforms from a pro-inflammatory state to an anti-inflammatory state[22]. At disease level, the atheroprotective role of KLF4 in ECs and monocytes/macrophages is demonstrated by increased atherosclerosis in ApoE-null mice with either EC-specific or myeloid-specific KLF4 ablation[20,23]. Given the protection that KLF4 brings about in the cardiovascular system, it is essential for us to explore how KLF4 can play a protection role in atherosclerosis, whether it works against the cholesterol-induced damage of foam cell-like phenotypic transformation in VSMCs. In this study, we discerned that foam cell-like phenotypic switching inhibited the function of KLF4 in VSMCs and thus a cholesterol damage protective role of KLF4 was exerted in atherosclerosis.
1 Materials and Methods
1.1 Reagents
Cyclodextrin (CD) cholesterol-complex and oil redO(ORO), were purchased from Sigma-Aldrich. Antibodies were purchased from suppliers as follows: anti-SM22α (ab14106), anti-LXRα (ab176323), anti-ABCA1 (ab18180), anti-CD68 (ab955), anti-NLRP3 (ab263899), anti-IL-6 (ab290735), anti-VCAM-1 (ab217572), anti-ACTA2 (ab124964), anti-KLF4 (ab214666), anti-phospho-p65 (Ser536) (ab76302), anti-IL-1β (ab254360), anti-β-Actin (ab6276), mouse IL-1β Elisa kit (ab241673), mouse IL-6 Elisa kit (ab222503) and mouse MCP-1 Elisa kit (ab208979) were from Abcam; anti-IL-10 (6026p-1-lg), anti-MCP-1 (66272-1-lg), anti-SREBP (28212-1-AP), anti-ICAM-1 (16174-1-AP), anti-HMGCR (13533-1-AP), anti-MCP-1 (66272-1-lg), anti-LGALS3 (60207-1-lg), anti-P65 (66535-1-lg), anti-MYD88 (67969-1-lg), anti-TLR4 (19811-1-lg), anti-LOX-1 (11837-1-AP), anti-CD36 (18836-1-AP) were from Proteintech; TRITC- or FITC-conjugated secondary antibodies were from KPL (Kirkegaard &Perry Laboratories, Inc). DyLight 649 AffiniPure goat anti-mouse antibody (E032610) was from EarthOx.
1.2 Cell culture and treatment
VSMCs were isolated from the thoracic aorta of wild-type (WT) andKLF4-/-mice and cultured in low glucose Dulbecco’s-modified Eagle’s medium (DMEM) (Invitrogen, US) supplemented with 20% fetal bovine serum (FBS, Gibco), 100 U/mL penicillin and 100 μg/mL streptomycin. The VSMCs were maintained at 37 ℃ in a humidified atmosphere containing 5% CO2, and only passages 4 to 15 cells at 70%-80% confluence were used in the experiments, except if stated otherwise.
Serum starvation was done by withdrawing serum and incubating the cells in 0.1% FBS for 24 hours before stimulated with cyclodextrin (CD) cholesterol-complexes (20 μg/mL) and transduction with adenoviruses as well as transfection with small interference RNA.
1.3 Adenovirus packaging and transduction
Full-length cDNA of KLF4 and PPARγ were cloned into pEGFP-C2, a mammalian expression vector and encoding a red-shifted variant of wild-type GFP, to generate pEGFP-KLF4 and pEGFP-PPARγ. Then the fusions of KLF4 and PPARγ plus GFP were subcloned into pAd/CMV/V5-DEST Gateway Vector (Invitrogen) to make the GFP tagged KLF4 and PPARγ adenovirus Ad-GFP-KLF4 and Ad-PPARγ, according to the manufacturer’s protocol. Ad-GFP was obtained the same to the above. All of these clones were verified by sequencing. The VSMCs were transducted with the above adenovirus (5×109pfu/mL) for 24 hours, washed and incubated in serum-free medium without adenovirus for 24 hours, then stimulated with or without cyclodextrin (CD)-cholesterol complex.
1.4 SiRNA transfection
The cultured VSMCs were grown to 50%-60% confluence, and then transfected with specific siRNA, siKLF4 (5′-CCAUUACCAAGAGCUCAUGCCACCC-3′), siPPARγ, or non-specific scrambled siRNA, siCon (5′-UUCUCCGAACGUGUCACGUTT-3′) using Lipofectamine iMax Reagent (Invitrogen) according to the manufacturer’s protocol. At 6-12-hour after transfection, VSMCs were treated with cyclodextrin (CD)-cholesterol complex as mentioned.
1.5 Western blotting
Lysates from cells were prepared with lysis buffer (1% Triton X-100, 150 mmol/L NaCl, 10 mmol/L Tris-HCl, pH 7.4, 1 mmol/L EDTA, 1 mmol/L EGTA, pH 8.0, 0.2 mmol/L Na3VO4, 0.2 mmol/L PMSF, and 0.5% NP-40). Equal amounts of protein (30-100 μg) were separated by 8% or 10% or 12% SDS-PAGE, and electro-transferred to a PVDF membrane. Membranes were blocked with 5% BSA (bovine serum albumin) for 2 hours at room temperature, and incubated with specific antibodies as described above at 4 ℃ overnight, and then with the HRP-conjugated secondary antibody (1∶10 000, Santa Cruz Biotechnology) for 1 hour. The blots were evaluated with the Chemiluminescence plus Western blot analysis kit or ECL (enhanced chemiluminescence) detection system. These experiments were replicated three times.
1.6 RNA isolation and quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was extracted from cell or tissue samples using TRIzol Reagent (Invitrogen) and treated with DNaseI to remove genomic DNA. A quantitative real-time reverse-transcriptase (RT)-PCR was performed with a Bio-Rad thermocycler and a SYBR green kit (Invitrogen) following manufacturer’s instructions. The relative mRNA expression was normalized to β-actin, using the 2-△△Ctmethod. The average threshold cycle for each gene was determined from at least three independent experiments (Table 1).

Table 1 Sequence-specific primers used in the experiments

Continued Table 1
1.7 Immunofluorescent staining
Cells were fixed in 4% paraformaldehyde and permeabilized with 0.1%-0.5%Triton X-100 at room temperature for 20 minutes. Thereafter, cells were incubated with anti-ACTA2 and anti-CD68 antibodies and further stained with appropriate TRITC- or FITC-conjugated secondary antibodies. Confocal microscopy was performed with the Confocal Laser Scanning Microscope Systems (Leica).
1.8 Cell viability assay using CCK8
VSMCs were seeded into 96-wells plates at a density of 2 000 cells/well and cultured with or without cholesterol for 72 hours. Afterwards, 10 μL of CCK8 solution was added to each well, and the cells incubated for 2 hours. The color intensity was measured at an absorbance of 450 nm (Tecan Infinite M200 microplate reader; LabX, Austria). All experiments were performed in triplicate and were repeated three times.
1.9 Cell adhesion
WT andKLF4-/-VSMCs were incubated with or without cholesterol complex for 72 hours as appropriate. Treated VSMCs were washed three times with PBS, and CELLTRACE Violet-labelled macrophages cells (1×106cells per well) were added to the VSMC cultures and incubated for 1 hour at 10 r/min at 37 ℃. Cells were then washed twice with PBS to remove non-attached cells. The VSMC layers with attached macrophages were fixed with 4% PFA, and adhered macrophages were measured using a fluorescence microscope at 50× magnification (DMI4000B).
1.10 Flow cytometry
The phenotype of the macrophage-like cells was analyzed by measuring the surface expression of the macrophage-specific marker CD68 using flow cytometry. After appropriate treatments, VSMCs were collected and stained with an anti-CD68 antibody (5 μL in 50 μL Perm/Wash buffer) or its isotype control (1 μL in 50 μL Perm/Wash buffer) incubated at 4℃ for 30 minutes in the dark. The stained cells were then washed and analyzed using the FACSCanto II system equipped with BD FACSDiva software (Becton-Dickinson, San Jose, CA, USA) to determine the percentage of CD68-positive cells.
1.11 Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using the PierceTM Agarose ChIP Assay Kit (Thermo Scientific) according to manufacture instruction. VSMCs were fixed in 1% formaldehyde for 10 minutes to cross link proteins with DNA. The cross-linked chromatin was then prepared and sonicated to an average size of 200-600 bp. The samples were precleared with Dynabeads Protein G (Life technology) for 30 minutes at 4 ℃. The DNA fragments were immunoprecipitated overnight at 4℃ with anti-KLF4 and normal rabbit IgG (Santa Cruz) antibodies. The precipitated DNA was recovered via phenol/chloroform extraction, and KLF4 binding site of PPARγ (Forward primer: 5′- TGG CAT CTC TGT GTC AAC CAT G-3′, Reverse primer: 5′- GCA TGG TGC CTT CGC TGA-3′) gene promoter region was amplified by qPCR. Each experiment was replicated at least three times.
1.12 Animals
TheKLF4-/-mice line which has a Cre-recombinase gene inserted into the endogenous KLF4 locus andApoe-/-mice were purchased from the Jackson Laboratory. All mice were housed in the temperature- and humidity-controlled facility, with standard laboratory chow (16% protein, 4% fat, 6% fiber) and ad libitum access to food and water.Apoe-/-andKLF4-/-animals were crossbred to obtainKLF4-/-Apoe-/-and theirKLF4+/+Apoe-/-littermates. To create atherosclerosis, 6-week-old maleKLF4-/-Apoe-/-and theKLF4+/+Apoe-/-mice were fed Western Diet containing 21% fat, and 1.5% cholesterol (D12079B from Research Diet). Eight weeks after Western Diet, the aortas were isolated to assess the extent and distribution of atherosclerotic lesions by Oil Red-Ostaining. Lesion area was measured with ImageJ and expressed as a percentage of the total area of aorta. Plasma levels of total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C) and triglycerides (TG) were determined using a kit from Nanjing Jiancheng Bioengineering Institution. All animal procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011) and was approved by the Institutional Animal Care and Use Committee of Hebei Medical University.
1.13 Oil red O (ORO) staining
Aortas and cultured VSMCs were washed three times with PBS, fixed for 20 minutes in 4% paraformaldehyde and stained for 30 minutes in 0.3% Oil RedO. The cells or aortas were then washed three times with PBS and photographed with microscope. Lesion areas were quantified with Image Pro Plus (IPP) software.
1.14 Lipid extraction and total cholesterol measurement
Aorta and intracellular total cholesterol was determined using a Cholesterol Quantitation Kit, according to the manufacturer’s instructions. VSMCs (1×106cells) were extracted with 200 μL of chloroform: isopropanol: octylphenoxy poly (ethyleneoxy) ethanol (IGEPAL CA-630) (7: 11: 0.1) in a micro-homogenizer. After centrifugation, the lipids were air-dried, vacuum-dried, dissolved and mixed until homogenous. The mixture was used to determine total cholesterol (μg/μL) using a colorimetric assay. Three independent experiments were conducted.
1.15 Thin Layer Chromatography (TLC)
TLC is a semi-qualitative analytical method that allows chemical compounds to be separated and partially identified in a complex mixture. Totally 30 μL lipid solution extracted from aortas of WT andKLF4-/-mice fed Western diet were put in TLC plate (silica gel 60 F-254, Merk, Germany) and were separated using dichloromethane/methanol (97∶3V/V) and dichloromethane/ethyl acetate (90∶10V/V) as solvent systems for two-dimensional chromatography.
1.16 Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad Software Inc., La Jolla, CA, USA). Data from at least three independent tests performed in triplicates are presented as the means ± SEM. Error bars in the scatterplots and the bar graphs represent SEM. Normality test and variance homogeneity test on the data were conducted firstly. For data with normal distribution, ANOVA was employed for statistical analysis when more than two groups were compared, and a two-tailed Student’sttest was used when comparison between two groups was performed. Nonparametric Kruskal-Wallis rank sum test was used for continuous variable with non-normally distributed data. In all cases, statistical significance was concluded where the two-tailed probability was less than 0.05. One representative experimental result was selected to display in the present study from three or more independent experiments.
2 Results
2.1 Depletion of KLF4 contributes to the development of atherosclerosis in mice
To evaluate the sensitivity ofKLF4-/-mice to atherosclerosis,KLF4-/-Apoe-/-mice were made by hybridization ofKLF4-/-andApoe-/-mice.Apoe-/-andKLF4-/-Apoe-/-mice were both fed with Western Diet for 8 weeks. A more significant positive oil redO(ORO) staining was observed in aortic section (Fig.1A) and aortic sinus (Fig.1B) as well as aortic cross-section (Fig.1C) ofKLF4-/-Apoe-/-mice compared to that ofApoe-/-mice. Meanwhile, the diet significantly increased aortic total cholesterol (TC) and cholesteryl ester (CE) content in artery wall ofKLF4-/-Apoe-/-mice (Fig.1D and E). Moreover, the levels of serum TC, low-density lipoprotein cholesterol (LDL-C) and triglyceride (TG) gradually increased and were significantly higher inKLF4-/-Apoe-/-mice than that inApoe-/-mice (Fig.1F-H). Overall, these data indicated thatKLF4 depletion may contribute to increased susceptibility to atherosclerosis in mice.
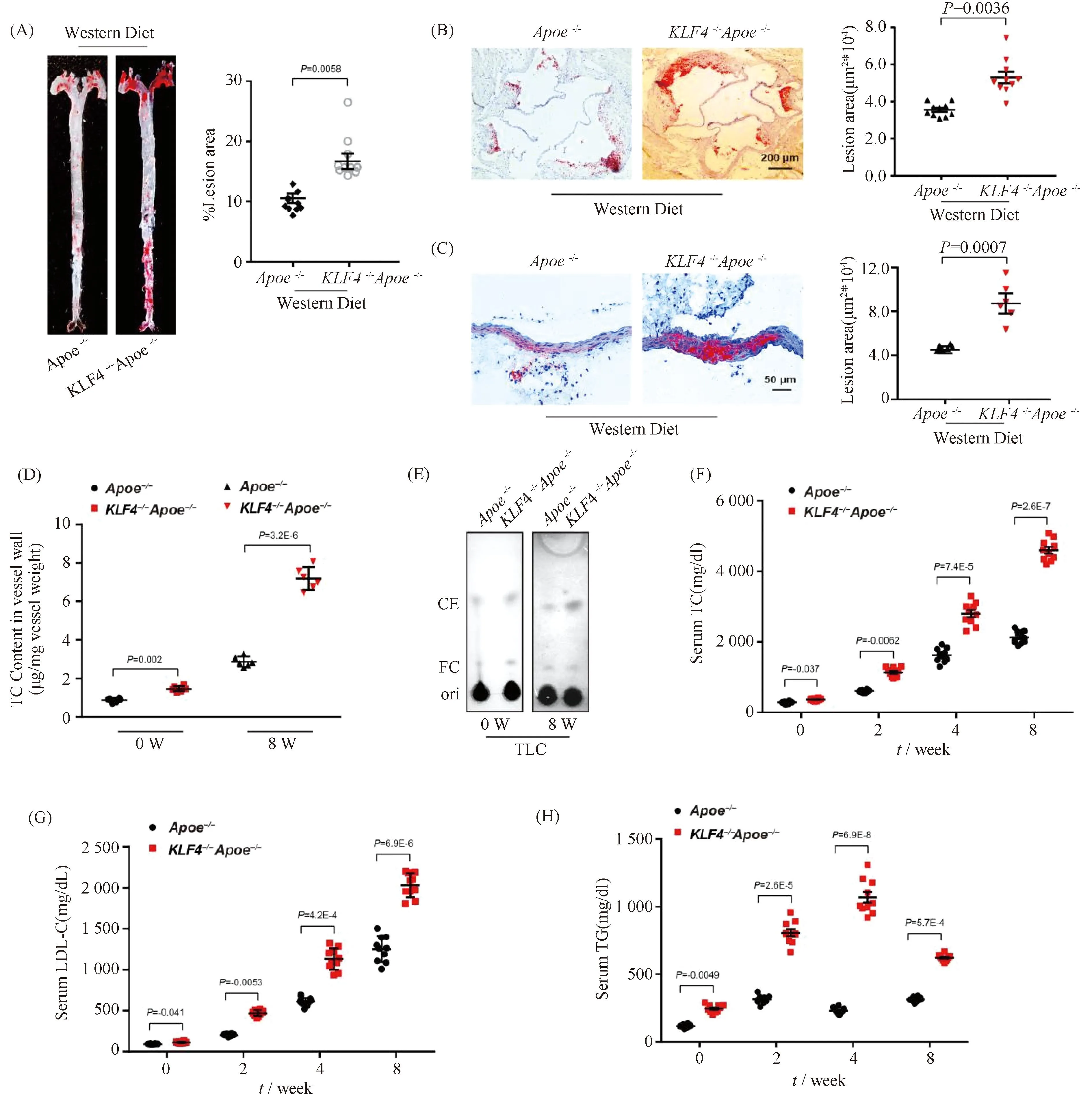
Fig.1 Impaired KLF4 expression is associated with development of atherosclerosis Apoe-/- (n=10) and KLF4-/-Apoe-/- (n=10) mice were fed with Western Diet for 8 weeks, and then aortas and aortic sinus were captured. Aortas, aortic sinus and artery wall were stained with ORO and cholesterol content of artery wall and serum were measured. (A-C) Representative images of en face ORO-stained aortas (A), aortic sinus (B), aortic cross sections (C) and quantification of lesion areas are shown. (D) TC content analysis for aortic wall of Apoe-/- and KLF4-/-Apoe-/- mice fed Western Diet (n=6). (E) TLC analysis for cholesterol content in aortic wall of Apoe-/- and KLF4-/-Apoe-/- mice fed Western Diet (n=6). FC: free cholesterol; CE: cholesterol ester. (F-H) Serum TC (F), LDL-C (G) and TG (H) levels of Apoe-/- and KLF4-/-Apoe-/- mice fed Western Diet were monitored (n=10). Images are representative of at least three independent experiments. The data were presented as the mean ± SEM and statistical significance was assessed by Shapiro-wilk to test normality. Data in (A), (B) and (C) were analyzed by one-way ANOVA. Data in (D), (F), (G) and (H) were analyzed by two-way ANOVA
2.2 Cholesterol promotes foam cell-like VSMCs formation
Normally, VSMCs maintain contractile phenotype in the resting state and undergo phenotypic transformation in certain stimulation. To explore the effects of cholesterol loading on VSMCs phenotypic transformation, we first examined various VSMCs functional and phenotypic parameters including cell viability, lipid deposition and foam cell markers expression. CCK-8 assay was utilized to detect the viability of VSMCs under cholesterol stimulation of different concentrations. Data showed that there were no significant differences of cell viability among VSMCs treated with cholesterol in concentration of lower than 20 μg/mL while a sharp decrease of cell viability was detected in VSMCs stimulated by cholesterol above 20 μg/mL which means a clear toxic effect of cholesterol was generated (Fig.2A). Consistent with previous reports[24], lipids accumulation was found in VSMCs incubated with cholesterol of 10 μg/mL for 72 hours by ORO staining (Fig.2B), indicating the foam cells were formed. To further characterize the phenotypic conversion of VSMCs, qRT-PCR was used to assess the expression of contractile phenotype markers, such asACTA2 andSM22α, and foam cell-like phenotype markers including CD68 and LGALS3. Results showed a significant increase in macrophage-related gene expression while a decline in the expression of contractile phenotype-related gene (Fig.2C). These data may suggest that VSMCs altered to a macrophage-like phenotype after cholesterol loading.
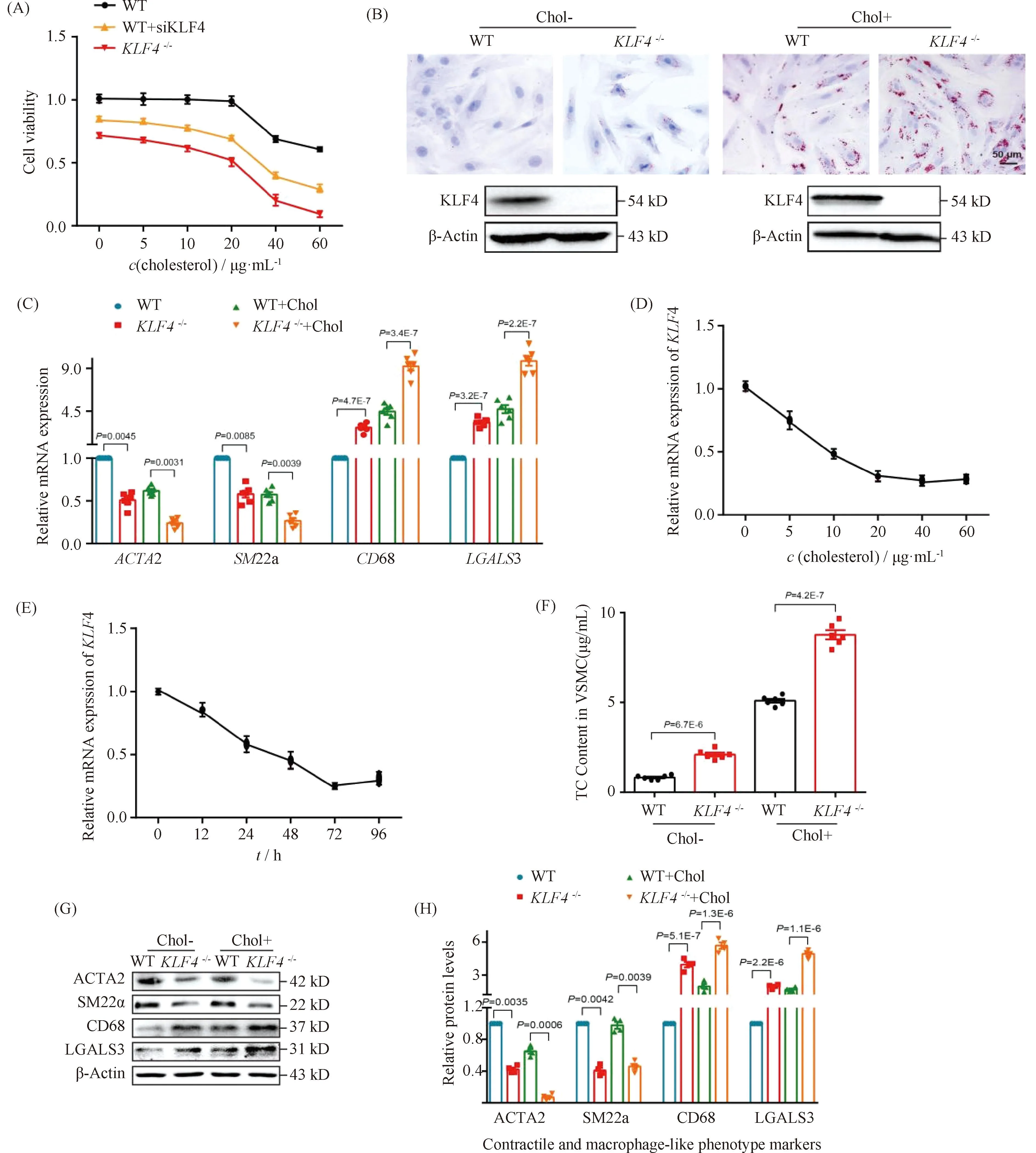
Fig.2 Loss of KLF4 promotes foam cell-like VSMCs formation WT and KLF4-/- VSMCs were incubated with/without cholesterol, CCK8 assay, ORO staining, qRT-PCR, TC content detection and Western blot were used to test cell viability, foam cells formation, phenotypic marker and cholesterol content respectively. (A) CCK8 assay were used to detect cell viability of WT, WT transfected with siKLF4 and KLF4-/- VSMCs with cholesterol stimulation of different concentration (0, 5, 10, 20, 40, 60 μg/mL) for 72 hours (n=6). (B) ORO staining of WT and KLF4-/- VSMCs stimulated with/without cholesterol (72 hours) (n=6). (C) Relative mRNA level of contractile (ACTA2, SM22α) and foam cell-like phenotypic markers (CD68, LGALS3) in WT and KLF4-/- VSMCs with/without cholesterol incubation for 72 hours (n=6). (D) Relative mRNA expression of KLF4 under the stimulation of cholesterol in different concentration (0, 5, 10, 20, 40, 60 μg/mL) (n=6). (E) Relative mRNA expression of KLF4 under the stimulation of cholesterol for different times (0, 12, 24, 48, 72, 96 hours) (n=6). (F) TC content analysis for WT and KLF4-/- VSMCs with/without cholesterol incubation (72 hours) (n=6). (G-H) Relative protein (G) level and quantification (F) of contractile (ACTA2, SM22α) and foam cell-like phenotypic markers (CD68, LGALS3) in WT and KLF4-/- VSMCs with/without cholesterol incubation for 72 hours (n=6). Images are representative of at least three independent experiments. The data were presented as the mean ± SEM and statistical significance was assessed by Shapiro-wilk to test normality. Data in (A) was analyzed by two-way ANOVA. Data in (C), (D) and (F) were analyzed by unpaired t test
2.3 KLF4 mitigates cholesterol induced foam cell-like phenotype switching in VSMCs
To further explore the way of cholesterol regulates KLF4 expression, qRT-PCR were used to detectKLF4 expression in condition of different dose and different time of cholesterol stimulation. We found thatKLF4 expression was significantly dose- and time-dependent (Fig.2D-E). Next, to investigate the role of KLF4 in the progress of cholesterol induced foam cell-like phenotype formation, WT VSMCs transfected with siKLF4 andKLF4-/-VSMCs were used to incubate with cholesterol of different concentrations and then to detect parameters reflecting VSMCs functions including cell viability, lipid deposition and phenotypic markers of contractile and foam cell-like. Results of CCK-8 assay showed that an obvious decline of cell viability was detected both in siKLF4 andKLF4-/-VSMCs in the treated condition of over 15 μg/mL cholesterol, suggesting that the toxic effect of cholesterol was moved up owing toKLF4 deletion (Fig.2A). ORO staining also displayed a severer lipids sedimentation due to the disruption of KLF4 (Fig.2B). TC content measurement showed a remarkable climb of TC storage inKLF4-/-VSMCs compared to those of WT controls even though in the basal state without cholesterol treatment, and this climb could be amplified by cholesterol stimulation (Fig.2B and F). qRT-PCR and Western blot of phenotypic markers indicated an up-regulation of foam cell-like markers along with a down-regulation of contractile phenotype markers inKLF4-/-VSMCs with or without cholesterol incubation compared to WT VSMCs (Fig.2C, G and H). These data illustrated that KLF4 disturbance may promote VSMCs transform to macrophage foam cell-like phenotype.
To further identify a potential causative link between KLF4 expression and foam cell-like phenotype transformation in VSMCs,KLF4-/-VSMCs were transducted with Ad-GFP or Ad-GFP-KLF4. CCK-8 assay demonstrated that the downregulation of cell viability detected inKLF4-/-VSMCs was notably reversed by KLF4 rescue expression (Fig.3A). Moreover, decreased cholesterol deposition and TC content were also validated in Ad-GFP-KLF4-tranductedKLF4-/-VSMCs by ORO staining and TC quantification respectively (Fig.3B and C). QRT-PCR and Western blot showed that contractile phenotypic markers includingACTA2 andSM22αexpression were re-ascended and the expression of foam cell-like phenotypic markers such as CD68 and LGALS3 were declined inKLF4-/-VSMCs transducted with Ad-GFP-KLF4 compared toKLF4-/-VSMCs (Fig.3D-F). Meantime, immunofluorescence staining of ACTA2 and CD68 also validated this finding (Fig.3G). Furthermore, flow cytometry analysis showed thatKLF4-/-VSMCs under the condition of cholesterol treatment of 20 μg/mL for 72 hours induced 72.1% of VSMCs to present a foam cell-like phenotype, whereas only 23.0% of VSMCs adopted foam cell-like phenotype following pre-incubation with Ad-GFP-KLF4 and cholesterol (Fig.3H). Collectively, these findings suggest that KLF4 ameliorates foam cells-like phenotypic conversion of VSMCs.
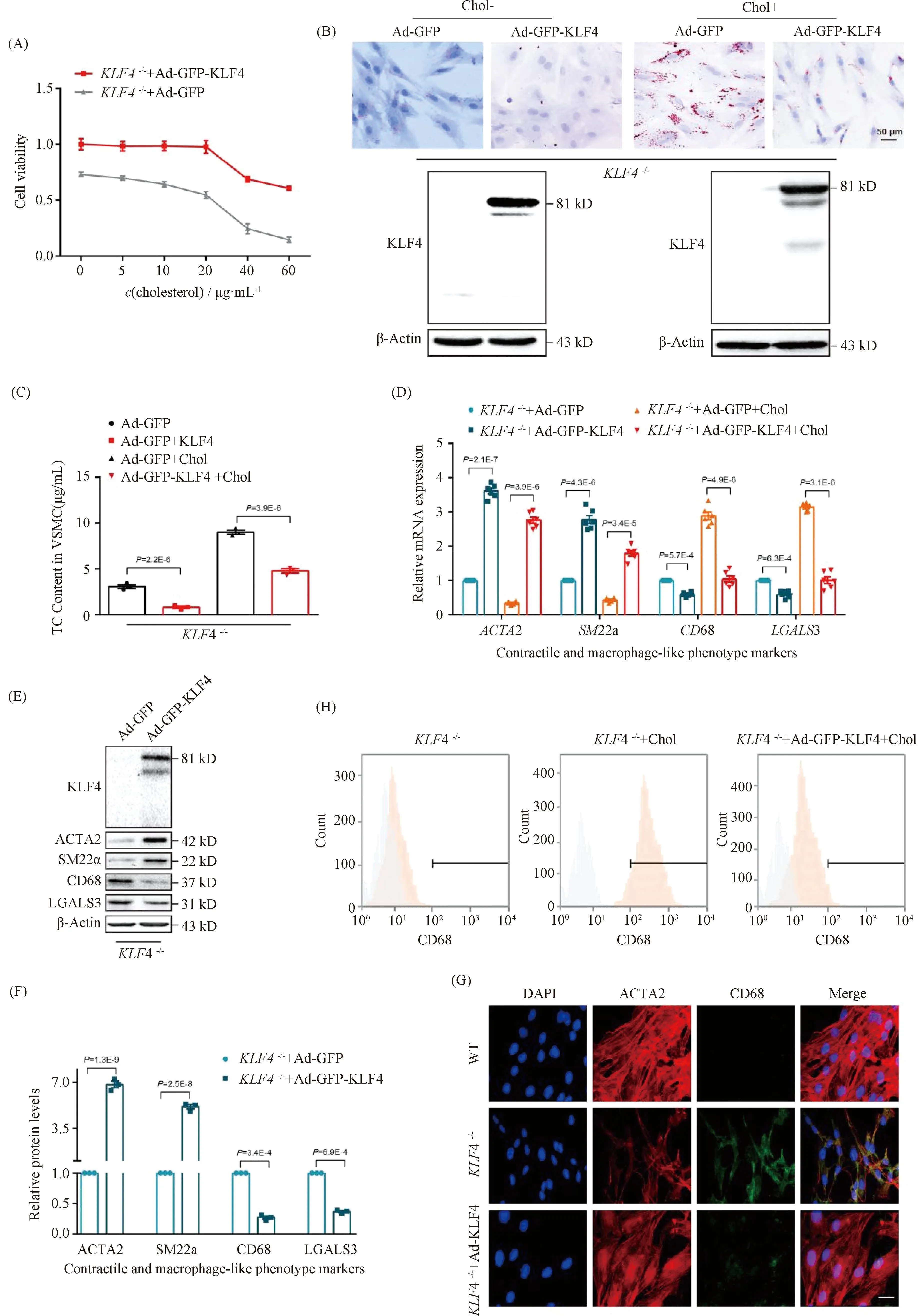
Fig.3 Rescue expression of KLF4 blocks the progression of foam cell-like VSMCs formation KLF4-/- VSMCs were transducted with Ad-GFP or Ad-GFP-KLF4 in the stimulation of cholesterol at different time points, then cells were harvested. CCK8 assay was used to detect cell viability (A), ORO staining was to marked foam cells (B), TC content analysis was utilized to suggest cholesterol content in VSMCs (C), qRT-PCR (D), Western blot (E), immunofluorescence staining (G) and flow cytometry (H) were used to indicate phenotype swithing (n=6). (D-F) Relative mRNA (D) and protein (E) levels and quantification (F) of contractile (ACTA2, SM22α) and macrophage-like phenotypic markers (CD68, LGALS3) in KLF4-/- VSMCs transfected with Ad-GFP or Ad-GFP-KLF4 in the condition of cholesterol incubation or not (n=6). (G) Immunofluorescence staining for ACTA2 and CD68 in WT and in KLF4-/- VSMCs transducted with Ad-GFP-KLF4 or not (n=6). Scale bar, 20 μm. (H) Representative flow cytometry analysis of CD68-positive cells in KLF4-/- VSMCs with or without cholesterol and Ad-GFP-KLF4 treatment. Images are representative of at least three independent experiments. The data were presented as the mean ± SEM and statistical significance was assessed by Shapiro-wilk to test normality. Data in (A) was analyzed by two-way ANOVA and data in (C), (D) and (F) were analyzed by unpaired t test
2.4 KLF4 suppresses foam cells-like phenotypic transformation via promoting cholesterol efflux in VSMCs
The abnormal deposition of cholesterol in artery wall and VSMCs contributes a lot in the pathogenesis and progression of atherosclerosis. To deeply investigate the mechanism of KLF4 inhibits foam cells-like phenotype formation, we first took advantage of the published RNA-sequencing (RNA-seq) analysis[16]and Chip-Sequence data[25]on endothelial cells (ECs)[16]and atherosclerosis lesion respectively. The data indicated that KLF4 overexpression affected the response to lipids (GO: 0033993) which was exhibited in the heat map in Fig.4A that genes associated with cholesterol efflux and oxidation were upregulated, whereas the cholesterol biosynthesis and uptake related genes were downregulated (Fig.4A). To validate this in VSMCs, we next detected the expression of cholesterol efflux and oxidation related genes, such asLXR,ABCA1,ABCG1,Ch25handCYP7A1 as well as cholesterol biosynthesis genes involvingSREBP2,HMGCR,PMVKandSQLE. QRT-PCR and Western blot revealed that compared to control group,KLF4 depletion decreased the levels of genes regulating cholesterol efflux and oxidation (Fig.4B, F and G) but heightened cholesterol biosynthesis genes expression (Fig.4C, F and G) and meanwhile these changes could be reversed by Ad-GFP-KLF4 transfected (Fig.4D-G). Overall, KLF4 can protect VSMCs from the damage of abnormal cholesterol accumulation through inhibiting cholesterol biosynthesis on the one hand and promoting cholesterol efflux and oxidation on the other hand, thereby the formation of foam cell-like VSMCs was prevented.
2.5 KLF4 alleviates cholesterol loading-induced VSMCs pro-inflammatory cytokines secretion and suppresses macrophage adhesion to VSMCs
Except for the deposition of cholesterol in artery wall and VSMCs, local pro-inflammatory factors in plaques and foam cells play a key role in atherosclerosis progression. In the data of the published RNA-seq analysis[16], the expression level of pro-inflammatory genes includingNLRP3,MCP-1,IL-6 andIL-1βwere significantly decreased owing to the transduction of Ad-GFP-KLF4 (Fig.5A). To clarify the potential mechanism of VSMCs-derived macrophage-like foam cells induced byKLF4 deletion in atherosclerosis, we also measured the levels of pro-inflammatory cytokines in cholesterol-overload WT andKLF4-/-VSMCs supernatants. Elisa assay indicated that the secretion of IL-6, IL-1β and MCP-1 were clearly increased by cholesterol accumulation both in WT andKLF4-/-VSMCs andKLF4 depletion amplified the increase of pro-inflammatory cytokines secretion (Fig.5B). Meanwhile, mRNA and protein levels of intracellular pro-inflammatory factors including NLRP3, MCP-1, IL-6, NF-κB, JNK as well as IL-1β were also examined, and similar results as supernatants that KLF4 disruption contributed to the expression of pro-inflammatory cytokines were obtained (Fig.5C and D). Furthermore, WT andKLF4-/-VSMCs were transducted with Ad-GFP-KLF4 and then were treated with or without cholesterol. Data showed that the raise of pro-inflammatory cytokines secretion was significantly reversed by KLF4 rescue expression not only in supernatants but also in cells (Fig.5D-F) but has no influence on cell viability (Fig.3A).
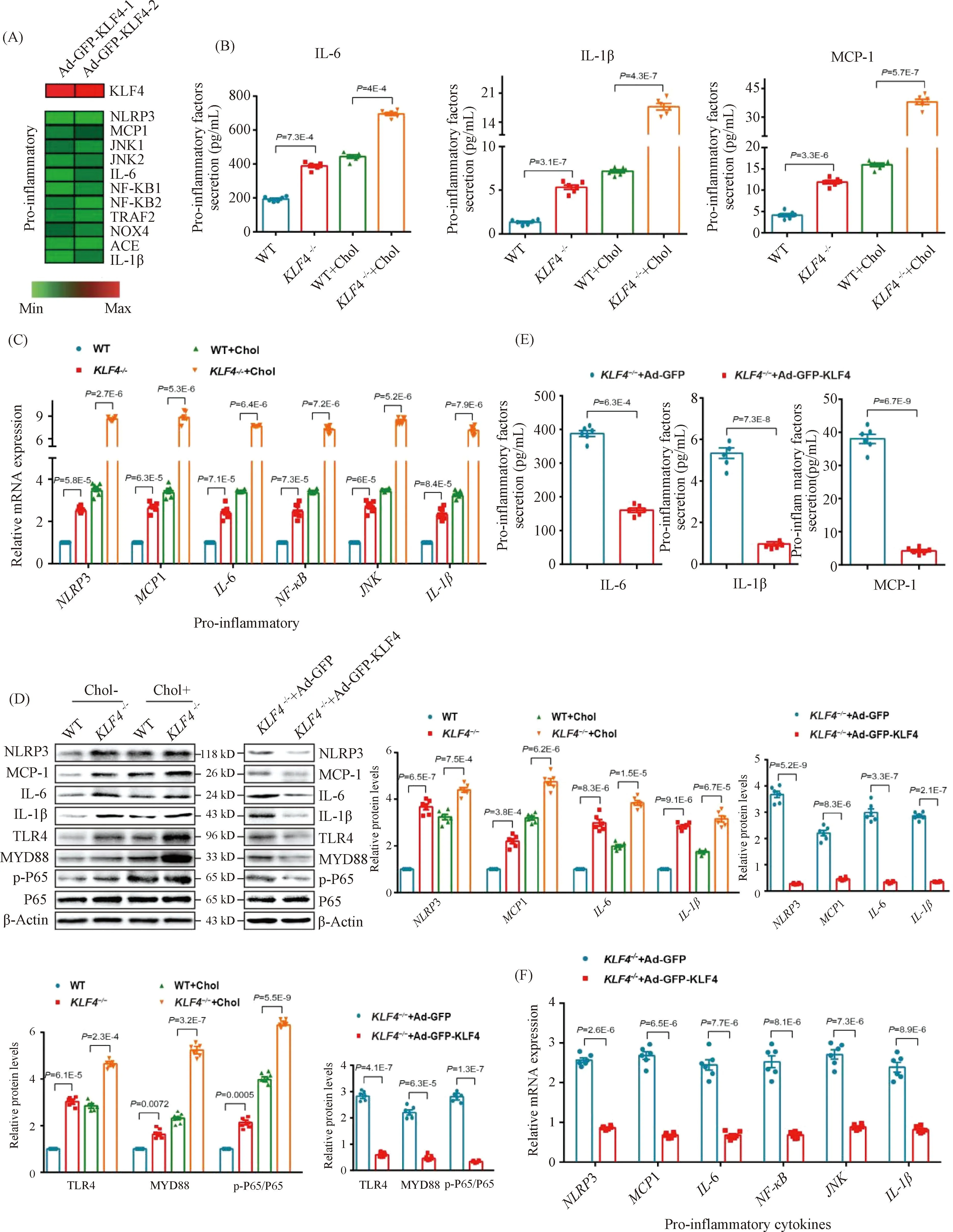
Fig.5 KLF4 inhibits secretion of pro-inflammatory factors (A) Heat map comparison of Ad-null versus Ad-GFP-KLF4 using log2 fold changes of pro-inflammatory genes. (B) Measurement of secretion of pro-inflammatory factors including IL-6, IL-1β and MCP-1 using Elisa kit in culture medium of WT and KLF4-/- VSMCs with/without cholesterol treatment for 72 hours (n=6). (C) Relative mRNA expression of genes regulating inflammation in WT and KLF4-/- VSMCs with/without cholesterol treatment for 72 hours (n=6). (D) Western blot and quantification of pro-inflammatory genes expression in WT and KLF4-/- VSMCs with/without cholesterol treatment for 72 hours and in KLF4-/- VSMCs transducted with Ad-GFP or Ad-GFP-KLF4 for 48 hours (n=6). (E) Measurement of secretion of pro-inflammatory factors (IL-6, IL-1β and MCP-1) using Elisa kit in culture medium of KLF4-/- VSMCs transducted with Ad-GFP or Ad-GFP-KLF4 for 48 hours (n=6). (F) Relative mRNA expression of genes regulating inflammation in KLF4-/- VSMCs transducted with Ad-GFP or Ad-GFP-KLF4 for 48 hours (n=6). Images are representative of at least three independent experiments. The data were presented as the mean ± SEM and statistical significance was assessed by Shapiro-wilk to test normality. Data in (B), (C), (D) were analyzed by two-way ANOVA, and data in (E) and (F) were analyzed by two-way ANOVA and unpaired t test
To further explore the effect of KLF4 disruption on the ability of VSMCs to recruit macrophages, cell adhesion assays employing CELLTRACE Violet-labelled Raw264.7 cells and VSMCs were used. We found a stronger adhesive ability to Raw264.7 in cholesterol loaded VSMCs rather than none cholesterol loaded VSMCs and this macrophages adhesion could be remarkably amplified byKLF4 depletion while reduced by pre-treatment of Ad-GFP-KLF4 (Fig.6 A and E). To further determine the mechanism by which cholesterol loaded VSMCs induced byKLF4 depletion recruited macrophages, qRT-PCR and Western blot were utilized to detect the expression of cell adhesion molecules including vascular cell adhesion molecule (VCAM)-1 and intercellular cell adhesion molecule (ICAM)-1. We showed that the expression of VCAM-1 and ICAM-1 were significantly increased inKLF4-/-VSMCs compared to WT VSMCs (Fig.6B-D), whereas KLF4 overexpression completely prevented this effect (Fig.6F-H). In summary, KLF4 plays a protective role in the process of macrophages adhering to cholesterol loaded VSMCs via blocking pro-inflammatory cytokines secretion.
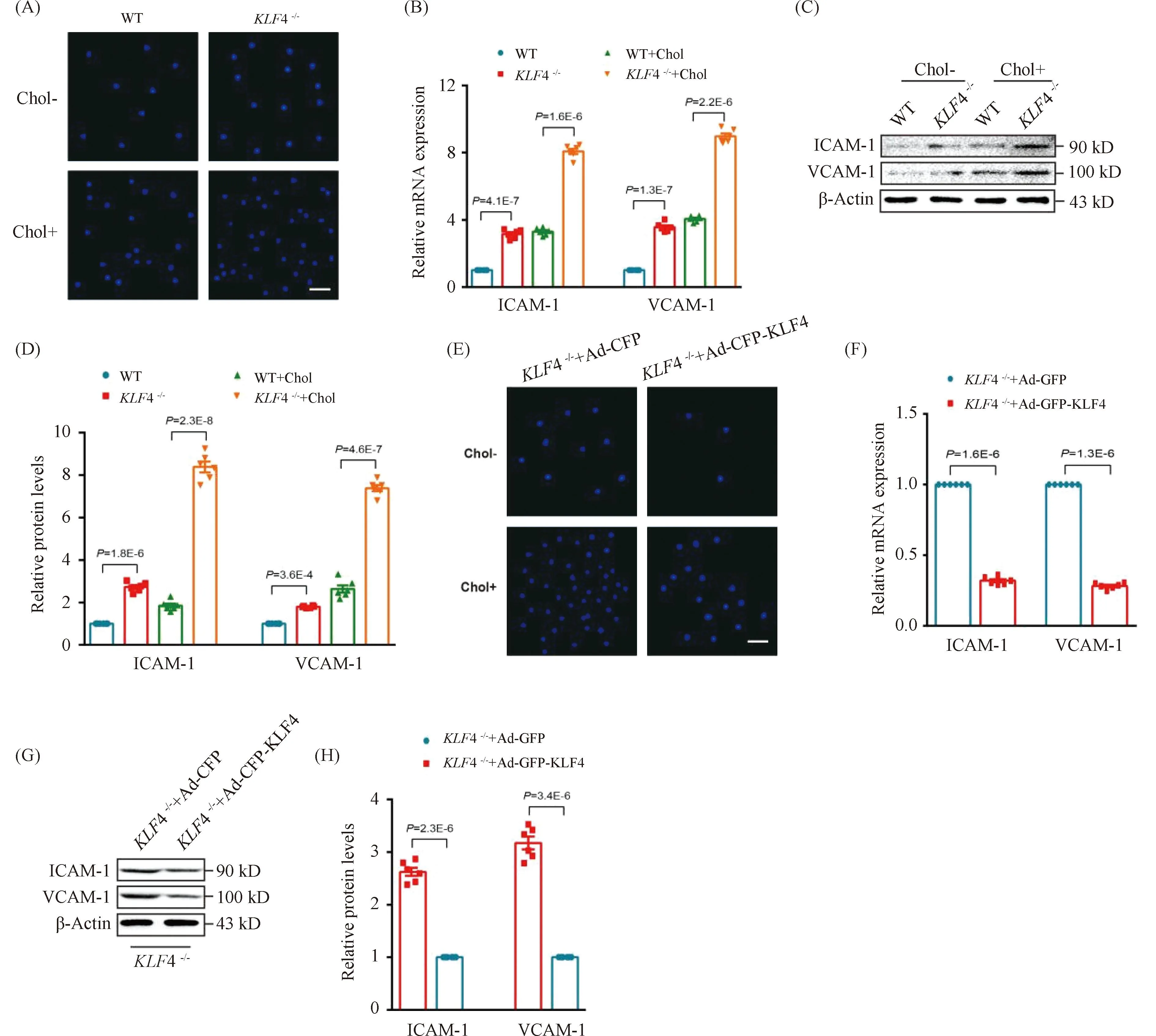
Fig.6 KLF4 alleviates cholesterol loading-induced macrophages adhere to VSMCs (A) Representative images of macrophages adhere to WT and KLF4-/- VSMCs with/without cholesterol treatment for 72 hours (n=6). Scale bar, 100 μm. (B) Relative mRNA expression of ICAM-1 and VCAM-1 in WT and KLF4-/- VSMCs with/without cholesterol treatment for 72 hours (n=6). (C-D) Western blot (C) and quantification (D) of ICAM-1 and VCAM-1 expression in WT and KLF4-/- VSMCs with/without cholesterol treatment for 72 hours (n=6). (E) Representative images of macrophages adhere to KLF4-/- VSMCs transducted with Ad-GFP or Ad-GFP-KLF4 with/without cholesterol treatment for 48 hours (n=6). Scale bar, 100 μm. (F) Relative mRNA expression of ICAM-1 and VCAM-1 in KLF4-/- VSMCs transducted with Ad-GFP or Ad-GFP-KLF4 for 48 hours (n=6). (G-H) Western blot (G) and quantification (H) of ICAM-1 and VCAM-1 expression in KLF4-/- VSMCs transducted with Ad-GFP or Ad-GFP-KLF4 for 48 hours (n=6). Images are representative of at least three independent experiments. The data were presented as the mean ± SEM and statistical significance was assessed by Shapiro-wilk to test normality, one-way ANOVA followed by Turkey’s test for post hoc comparison
2.6 KLF4 depletion modulates NF-κB and PPARγ pathways in VSMCs
To further find out the mechanism ofKLF4 deletion induced VSMCs dysfunction, we still use the published RNA-seq analysis[16]and Chip-Sequence[25]data and found that apart from the pro-inflammatory transcription factor NF-κB (Fig.4A), genes involved in the regulation of EC homeostasis such as PPARγ, eNOS and IL-10 were up-regulated due to the Ad-GFP-KLF4 transduction (Fig.7A). We next investigated the mRNA and protein levels of PPARγ, eNOS and IL-10 and found a sharp decrease of these genes inKLF4-/-VSMCs treated with/without cholesterol (Fig.7B and C) which could be reversed by Ad-GFP-KLF4 transduction (Fig.7D). PPARγ is a ligand-activated transcription factor that regulating numerous key cellular functions. Reports have shown that deficiency of PPARγ promotes phenotypic switching of VSMCs[26,27]. To further investigate whetherKLF4 deletion accelerated the transition of VSMCs from contractile to foam cell-like phenotype through inhibiting PPARγ and activating NF-κB pathway, the expression of VSMCs phenotypic markers, PPARγ and NF-κB pathway related cytokines were examined in WT andKLF4-/-VSMCs transfected with siCon and siPPARγ. PPARγ exhaustion evidently upregulated the expression ofNF-κBrelated genes (such asTLR4,MYD88 andP65 phosphorylation) as well as macrophage-like foam cells markers which was accompanied by a significant decline of contractile phenotype markers (ACTA2, SM22α) (Fig.7E and F). Compared toKLF4-/-controls, the levels of adhesion molecules (ICAM-1, VCAM-1), foam cells markers (CD68,LGLAS3,NF-κBrelated genes) were all lessened by Ad-PPARγ transfection while contractile phenotype markers, cholesterol efflux related genes (LXRα,ABCA1), and PPARγ expressions were boosted (Fig.7G and H). Collectively, we conclude that KLF4 protects VSMCs from contractile phenotype transforming to foam cell-like phenotype via inhibiting NF-κB pathway and maintaining normal PPARγ pathway.
2.7 PPARκ boosts KLF4 protective effect on VSMCs
To further validate the role of PPARγ in foam cells-like phenotypic switching ofKLF4-/-VSMCs, lipids accumulation and adhesion ability of macrophages to VSMCs were measured in WT andKLF4-/-VSMCs treated with siPPARγ. Treatment of siPPARγ not only notably subsided PPARγ (Fig.7E and F) expression but also promoted the damage effects ofKLF4 deletion as evidenced by the deposition of lipid droplets stained by ORO (Fig.8A) and TC content increment (Fig.8B) as well as the augmentation of adhesion ability of macrophages to VSMCs (Fig.8C). We next inspected if the above influences reduced by siPPARγ treatment could be reversed by the transduction of Ad-GFP-PPARγ. Results showed that PPARγ transduction could notably ameliorate lipids accumulation (Fig.8B and D) and macrophages adhesion ability to VSMCs inKLF4-/-VSMCs incubated with or without cholesterol (Fig.8E). To deeply confirm whether PPARγ is the downstream of KLF4, qRT-PCR and chip assay were conducted. qRT-PCR demonstrated thatPPARγsilencing could block the protective role of Ad-GFP-KLF4-mediated overexpression (Fig.8F) and chip assay suggested KLF4 could combine with PPARγ in WT VSMCs, whereas PPARγ could not be pulled down by KLF4 antibody inKLF4-/-VSMCs (Fig.8G). All of these experiments show thatPPARγis the target gene of KLF4 in VSMCs.
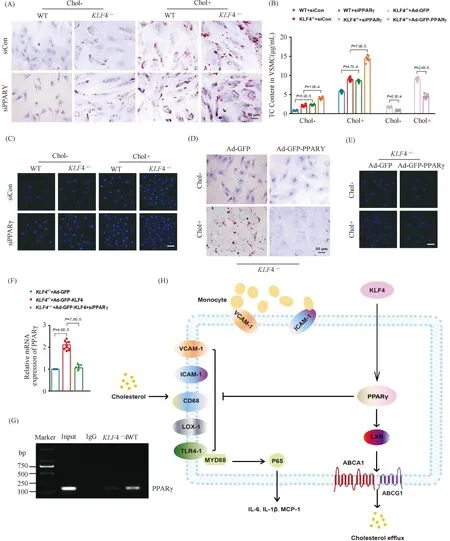
Fig.8 KLF4 blocks foam-like VSMCs formation through activating PPARγ (A) ORO staining of WT and KLF4-/- VSMCs transfected with siCon and siPPARγ for 48 hours and stimulated with/without cholesterol for 72 hours (n=6). (B) TC content analysis for WT and KLF4-/- VSMCs transfected with siCon and siPPARγ (for 48 hours) stimulated with/without cholesterol (for 72 hours) and in KLF4-/- VSMCs transfected with Ad-GFP or Ad-GFP-PPARγ (for 48 hours) (n=6). (C) Representative images of macrophages adhere to WT and KLF4-/- VSMCs transfected with siCon and siPPARγ with/without cholesterol treatment (n=6). (D) ORO staining of KLF4-/- VSMCs transducted with Ad-GFP or Ad-GFP-PPARγ stimulated with/without cholesterol (n=6). (E) Representative images of macrophages adhere to KLF4-/- VSMCs transducted with Ad-GFP or Ad-GFP-PPARγ stimulated with/without cholesterol (n=6). Scale bar, 100 μm. (F) A model showing the possible mechanism of KLF4-mediated protection of VSMCs against cholesterol-induced injury through PPARγ activation. Images are representative of at least three independent experiments. The data were presented as the mean ± SEM and statistical significance was assessed by Shapiro-wilk to test normality, one-way ANOVA followed by Turkey’s test for post hoc comparison
Overall, we concluded schematic representation of a working model in which KLF4 inhibits VSMC-derived foam cells formation by maintaining PPARγ normal function which inhibits activities of ICAM-1, VCAM-1, CD68, LOX-1 and expressions of target genes of NF-κB pathway and enhances cholesterol efflux and metabolism as well (Fig.8H).
3 Discussion
In the present study, we disclosed that KLF4 could protect VSMCs from cholesterol induced damages. KLF4 inhibited VSMCs-derived macrophage-like foam cells formation which characterized by diminished intracellular lipids deposition and changed biological functions. It has been reported that foam cells originated from VSMCs were observed in human atherosclerosis plaques[28]and cholesterol loaded VSMCs presented reduced contractile markers and raised foam cells markers indicating a phenotypic transformation to foam cells was made in VSMCs[11,24,29]. In addition, our results demonstrated that KLF4 disturbance accelerated this kind of phenotypic conversion in cholesterol-overload VSMCs and boosted various pro-inflammatory cytokines secretion involving IL-6, IL-1β and MCP-1 as well as promoted the adhesion of macrophages to VSMCs. It is prevailing to view that inflammation plays a critical role in almost all phases of atherogenesis, from initial recruitment of innate and acquired immune cells to infiltrate into arterial walls, to advanced thrombotic events[30,31]. Researches have confirmed the hypothesis that pro-inflammatory factors deficiency can both decrease and stabilize atherosclerotic plaques[32,33]. With respect to the pro-inflammatory cytokines secreted by cholesterol-loaded VSMCs, it is probably that VSMCs-derived macrophage-like foam cells might exert a central role in promoting plaque progress and if so restraining the dysfunction of VSMCs caused by cholesterol loading might be an effective way to inhibit atherosclerosis.Invitrodata of our research illustrated that KLF4 subsided the secretion of pro-inflammatory cytokines induced by cholesterol loading in VSMCs, suggesting that KLF4 may indeed be fruitful in impeding atherosclerosis.
Recruitment of macrophages into the aorta wall is regarded as an accelerant to the process of atherosclerosis[34]. The expression of ICAM-1 and VCAM-1 were up-regulated by cholesterol loading and meanwhile the adhesion ability of macrophages to VSMCs was heightened. KLF4 overexpression prevented these cholesterol-induced effects and thus revealing that KLF4 suppressed macrophages recruitment to VSMCs through the inhibition of ICAM-1 and VCAM-1 expression.
To deeply investigate the mechanism of KLF4 inhibits VSMCs-derived macrophage-like foam cells formation, we combined the published Chip-seq data of KLF4 from atherosclerosis lesion performed by Shankman et al. in 2015[25]and RNA-seq data performed by Li et al. in endothelial cells[16]and we detected target genes expression through qRT-PCR and Western blot. Although the RNA-seq data of Li. et al were specific to endothelial cells, they still have certain guiding significance in VSMCs and we conducted following experiments based on the two data base. Combining the two databases, results indicated that expression of a series genes associated with cholesterol uptake, efflux, macrophages and inflammation were influenced by KLF4.
Researches have shown that improving cholesterol efflux accelerates foam cell-like VSMCs to restore contractile phenotypes and it may contribute to the formation of VSMC-containing fibrous capsinvivo[24]. Accordingly, preventing cholesterol accumulation through stimulating cholesterol efflux will protect VSMCs from cholesterol injury and benefits to plaque stability. A series of genes were involved in cholesterol uptake and efflux, such asLOX-1,CD36,TLR4,LXR,ABCA1 andABCG1.LOX-1 andCD36, specific receptors for ox-LDL, is important for atherogenesis through up-regulating lipid deposition[35]. Likewise, TLR4 is also a contributor in early atherogenesis by promoting foam cells formation[36]. The nuclear transcription factor LXR plays a significant role in both lipid metabolism and inflammation[37]. ABCA1 and ABCG1 are ABC transporters which play critical roles in reverse cholesterol transport (RCT) and anti-plaque formation[38,39]. Interestingly, we found thatKLF4 silence lead to an up- and down-regulation of proteins related to cholesterol uptake and RCT respectively in VSMCs foam cell-like state. KLF4 transfection significantly down-regulated LOX-1, CD36, and TLR4 levels in VSMCs with or without cholesterol loading, whereas further increased LXR, ABCA1 and ABCG1 expressions. Moreover, ORO-marked lipid drops were significantly dwindled in Ad-GFP-KLF4 pre-treated VSMCs consistent with the results of intracellular cholesterol quantification, which indicating KLF4 effectively inhibits cholesterol loading. Since VSMCs were not treated with apolipoprotein A-I (ApoA-I) to promote cholesterol efflux, the decreased cholesterol levels in cholesterol-loaded VSMCs transfected with KLF4 likely occurred as a result of inhibition of cholesterol intake through LOX-1 and TLR4 downregulation.
NF-κB, a transcription factor, is linked to the activation of inflammatory pathway and the onset of atherosclerosis[40]and controls the expression of ICAM-1 and VCAM-1 in atherosclerotic lesions[41]. Our data demonstrated that NF-κB pathway participates in the process of KLF4 disruption induced cholesterol-overload VSMCs injury. ICAM-1 and VCAM-1 levels were both amplified inKLF4-/-VSMCs with cholesterol loading and this amplification could be significantly reversed by Ad-GFP-KLF4 pre-treatment. Furthermore, KLF4 overexpression significantly decreased the percentage of CD68-positive VSMCs and inhibited ACTA2 decline after cholesterol loading demonstrating the protective effects of KLF4 in foam cell-like phenotypes transformation of VSMCs.
PPARγ has been searched extensively as therapeutic targets for CVD over the past few decades[42,26]. PPAR-γ exerts a significant role in controlling vascular homeostasis and its dysfunction may lead to atherosclerosis, restenosis, and hypertension[43]. However, the underlying molecular mechanism mediating these effects remains ambiguous. In atherosclerotic lesions, PPAR-γ are highly expressed and its activation improves inflammatory effects in cardiovascular cells[44]. In endothelial cells, PPAR-γ leads to upregulation of NOS, increasing the bioavailability of the nitric oxide (NO)[45]. In VSMCs, PPARγ may limit cellular proliferation through changes in telomerase activity[46-48]and also in the expressions of retinoblastoma protein (Rb) and c-fos, and prevent cellular migration through enhanced transcription factor Ets-1[49, 50]. Furthermore, PPARγ can also promote cholesterol efflux and reverse cholesterol transport in macrophages[51]. To investigate whether PPARγ has an effect on KLF4 ablation induced foam cell-like VSMCs formation, we first demonstrated the relationship of KLF4 and PPARγ that overexpression of KLF4 enhances PPARγ activation, whereasKLF4 deletion reduced PPARγ activation. Consistent with this, KLF4 transfection significantly down-regulated LOX1, CD36, SR-A and increased LXR, ABCA1 and ABCG1 expressions. Reports have shown that PPARγ[42]as well as LXR[52]prevent the activation of NF-κB, consistent with our results that TLR4, MYD88, p-P65, ICAM1 and VCAM1 expressions were diminished in KLF4 transfected VSMCs.PPARγsilence abolished KLF4-induced ACTA2 enhancement, LOX1 decrease, NF-κB relevant proteins decline and upregulation of LXR, ABCA1 and ABCG1. These results point out that PPARγ activation is vital to protect VSMCs from cholesterol-induced damages induced by KLF4 ablation.
In a word, our data suggest that cholesterol loading of VSMCs caused byKLF4 depletion elevates inflammatory factors levels and macrophages adhesion ability, which may increase lipids burden in plaque. KLF4 inhibits cholesterol uptake and ameliorates damages of foam cell-like phenotypic conversion induced by cholesterol overload in VSMCs and it appears to be a promising drug to treat atherosclerosis.
