
高通量测序技术在低频突变检测中的应用
2024-02-22 09:37:30栾洋尤馨悦杨劲
遗传 2024年2期
栾洋,尤馨悦,杨劲
综 述
高通量测序技术在低频突变检测中的应用
栾洋1,尤馨悦1,杨劲2
1. 上海交通大学医学院公共卫生学院,上海 200025 2. 中国药科大学药学院,南京 210009
体细胞突变的累积与衰老、肿瘤及多种疾病的发生密切相关。在正常组织细胞中,基因组中自发突变和诱发突变的变异等位基因频率极低,对这类低频突变的检测一直面临挑战。第二代和第三代高通量测序(next-generation sequencing,NGS)技术的出现,可以实现任意物种全基因组上变异的直接检测,克服传统突变检测技术的诸多局限性。但是常规NGS由于测序错误率较高从而限制了其在低频突变检测上的应用,基于分子一致性测序策略进行错误矫正的高准确性NGS测序技术作为有效的低频突变检测工具,有望在环境诱变剂的评价与研究、细胞与基因治疗药物风险评估、人群健康风险监测和生命科学基础研究领域发挥重要作用。本文对比经典突变检测方法,对基于NGS的低频突变检测技术研究进展进行综述,并对应用前景进行展望,以期为该技术的进一步开发、研究和在相关领域的应用提供参考。
高通量测序技术;低频突变;分子一致性测序;致突变;风险评估
基因突变是指基因组中遗传物质发生的永久性改变。突变可因DNA复制或修复错误等自发产生,也可被外源性因素如辐射、化学物质、病原体、基因编辑和病毒载体转入等诱导产生。发生在生殖细胞上的突变能够传递给下一代,与遗传性疾病的发生和进化有关;而体细胞上的突变虽不会遗传,但与衰老、癌症的发生发展以及神经退行性疾病和心血管疾病等多种疾病密切相关[1~6]。因此,对突变进行高效、准确的检测是实现人群风险监测及预警的关键途径。过去几十年间,一系列依赖于特定的报告基因、基于表型检测的突变检测方法已得到充分验证,并被收入到相关毒性测试指南中,广泛应用于化学物致突变性评价。然而,上述方法大多需要使用特殊的细胞或者转基因动物模型,欠缺普适性,并且基于报告基因的方法有可能产生结果的偏倚。此外,致突变因素的暴露虽然会引起基因突变频率的升高[7],但是在细胞表型和功能未发生变化时,仍难以被现有技术检测到。以肿瘤的发生为例,只有当关键基因的癌基因或抑癌基因发生突变(驱动基因突变)时,才会使受到影响的细胞生长失去控制,细胞因此获得增殖优势形成克隆,从而最终发展为肿瘤(图1A)。而绝大多数位点的突变(中性突变)并不能让细胞获得选择性的生长优势。因此,早期阶段的细胞即使基因组突变频率有所升高,但尚未被正向选择因此在细胞群中占比很低,就难以被经典的基于表型的突变检测方法检测到。
第二代和第三代高通量测序(next generation sequencing,NGS)技术可对任意物种、组织在全基因组上进行无偏倚测序,使得突变的直接检测成为可能。但是,目前普遍使用的NGS测序错误率很高,达到千分之几,因此会掩盖掉低频发生的突变。肿瘤作为恶变细胞的克隆,相同的突变存在于多数样本细胞中,测序错误率并不影响结果判断。因此,现行NGS技术多应用于遗传性基因改变的体细胞、肿瘤细胞以及体外诱导干细胞克隆或单细胞上的突变分析。然而,当正常体细胞受到致突变因素作用后,全基因组上发生突变负荷的增加则难以通过现行NGS检测,因为在人类()和小鼠()样本中,检测到的自发突变频率约为10–9~10–7,即平均每107~109碱基对中才会出现1个突变[8~11]。细胞受到诱变作用后,突变频率即使增加几百倍,差别仍然会被在~10–3数量级的测序错误率所掩盖。同样,早期阶段时,细胞群体里仅有少数细胞发生恶性转化,以这样异质细胞混合体来源的DNA作为测序样本,其低频度发生的突变也难以通过现行NGS检测到。因此,只有开发错误率足够低的测序技术,才能实现体细胞基因组上低频突变的检测[12](图1B)。低频突变的检测一直是相关领域的技术难点,但作为研究热点近年来也取得了快速进展。本文着重针对环境诱变剂和药物风险评估研究领域,概括介绍传统突变检测技术,并就近年来基于NGS的低频突变检测方法进展进行综述,并对其应用和前景进行展望。
1 传统的低频突变检测方法
1.1 基于表型检测的基因突变实验
传统的基因突变实验多基于报告基因进行检测,通过分析基因组上内源性或者外源性报告基因产生的可检测表型变化来反映报告基因的突变情况。通常利用报告基因突变的细胞数目比例计算突变频率,还可借助PCR方法扩增突变细胞内的报告基因序列,结合PCR产物测序获得突变特征。常见的基因突变实验包括经典的基于细菌模型的细菌回复突变实验(Ames实验),利用哺乳动物细胞模型进行检测的基因突变实验、基因突变实验和基因突变实验等;利用转基因动物模型(如Big Blue大鼠/小鼠、MutaMouse小鼠和delta转基因大鼠/小鼠等)的体内基因突变实验,如基因突变实验、基因突变实验和基因突变实验等。此外还有近年来发展起来的外周血/基因突变实验[13~18]。/基因位于X染色体上,编码了细胞表面的糖基磷脂酰肌醇锚定蛋白,该基因发生突变时,锚定蛋白连接的细胞表面分化抗原会缺失,利用流式细胞术可以实现快速检测,从而推算/基因突变频率[15],该实验目前已用于啮齿类动物致突变性评价和人群风险评估。

图1 普通NGS对肿瘤细胞不同进展阶段和低频突变的检测能力
A:细胞发生恶性转化与不同阶段的检测技术。B:传统NGS难以检测体细胞超低频突变。
基因突变实验已被列入经济合作与发展组织(Organisation for Economic Co-operation and Development,OECD)化合物遗传毒性测试指南[19~22]、国际人用药品注册技术要求国际协调理事会(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)M7指导原则和我国各类化学品安全性评价的指南、程序或指导原则中。其中,Ames实验是最经典也是应用最为广泛的突变检测方法,具有较高灵敏性,通常在化学物致突变性的早期评价中使用。利用哺乳动物细胞进行的体外基因突变实验操作简单,但由于体外代谢活化系统与人体和其他生物体内代谢活化系统的差异并不能完全或真实反映化学物在体内代谢条件下的毒性特征以及靶器官的特异性。转基因动物体内基因突变实验能够克服体外实验的不足,可对转基因动物任意组织器官的突变进行检测,但是其检测需要将报告基因从基因组上切割并包装为噬菌体,再转染至细菌中,最终在细菌中检测报告基因的突变情况[17,21,23],实验操作繁琐、成本高、实验周期较长。啮齿类动物外周血基因突变实验比转基因动物实验操作简便,并且可以整合到一般毒性实验中,因此开始被广泛使用[24]。但由于使用的是外周血红细胞,因此只能反映骨髓造血器官的基因突变频率,并且因为红细胞无法获得DNA从而无法进行测序以考察突变特征。
上述基于内源或外源性报告基因的基因突变实验结果可能存在偏倚,因为报告基因不能代表全基因组,并且因同义突变不改变表型从而导致突变频率可能被低估;此外,对片段长度较短的报告基因分析突变特征也不能反映突变在全基因组位点、侧翼序列和链的倾向性。最为关键的是,借助外源报告基因的体内突变检测方法只能依赖于特殊的转基因动物模型,并不能直接用于一般人群或普通动物基因突变的检测。
1.2 基于基因型筛选的突变检测方法
基于基因型筛选的分子生物学方法也可用于低频突变的检测,包括限制性片段长度多态性/聚合酶链反应技术(PCR-RFLP)[25]、随机突变捕获技术(random mutation capture,RMC)[8]和微滴式数字PCR技术[26,27]等。前两者利用限制性内切酶酶切位点在突变前后经限制性内切酶酶切效果的不同进行突变的检测,后者通过对目标序列模板在单分子水平逐一进行PCR反应,从而对目标序列上的突变进行检测。与基于表型筛选的基因突变实验不同,这类方法直接对碱基序列进行分析,可以检测到108个碱基对中的1个突变,准确性较高[8,25]。但上述方法可检测的突变位点有限,PCR-RFLP和RMC只能对限制性内切酶酶切位点的突变进行检测,微滴式数字PCR主要用于对已知的突变进行定量检测,并且方法检测通量均较低。因此,与基因突变实验类似,上述方法仍然不能完全反映真实状态下全基因组水平的突变情况以及突变的序列偏向性。
综上所述,无论是基于表型筛选的基因突变实验还是基于基因型筛选的分子生物学检测方法,尽管准确性较高,能够实现达到自发突变水平的低频突变的检测,但由于对报告基因或序列的依赖,都会导致分析结果存在偏倚,也很难实现任意物种和任意组织细胞的全基因组水平的突变检测,限制了其在更为一般的低频突变检测场景的应用。
2 NGS应用于低频突变检测
1990年启动的人类基因组计划(Human Genome Project,HGP)正式开启了应用测序技术解析基因组序列、探究生命奥秘的大门。在HGP 计划完成之后出现的NGS技术,革命性地推动了基因研究领域的发展,实现了自动化、高通量和低成本检测,使其成为目前生命科学研究的常规手段,广泛应用于宏基因组学、进化研究、产前诊断和肿瘤研究等领域。NGS技术可对任一物种或组织细胞全基因组的序列进行高通量检测,直接获得核苷酸序列的变异信息,因此有可能直接对外源致突变剂诱导的突变进行定性和定量分析。理论上,NGS可以克服传统的突变检测手段对报告基因或检测位点的依赖性,是一种非常有潜力的手段,但因为常规NGS的随机测序错误率约为10–3~10–2[28,29],这样高的测序错误率限制了其在低频突变检测中的应用。NGS测序前样本采集和DNA提取、文库制备过程过程中的DNA片段化和DNA扩增、测序中的碱基识别过程以及测序后的生物信息学分析过程都有可能引入错误,从而影响突变检测的准确性[30~35]。千分之几的测序错误率不会影响NGS检测胚系突变和本质上属于细胞克隆的肿瘤细胞基因突变,通过提高NGS的测序深度仍然可以获得可信的变异信息。但是,正常体细胞突变的发生率及变异等位基因频率(variant allele frequency,VAF)很低,Lynch等[8]报道哺乳动物细胞每分裂一次,自发突变发生的概率约为10–9~10–8。在人类和小鼠样本中,检测到积累的自发突变频率约为10–9~10–7,即平均每107~109碱基对中才会出现1个突变[9~12]。在肿瘤高发的Tg-rasH2转基因小鼠模型中,即使给予致突变剂处理后,基因上绝大多数突变的VAFs值也只达到~10–4水平,且仅在单个DNA片段中被检测到[36]。相比于这些驱动基因上的诱发突变,正常组织细胞中自发的体细胞突变其VAFs值可能会更低,甚至仅在少数几个细胞中存在。也就是说,对于每个位点而言,其由于测序错误导致的变异的比例约为0.1%~1%;当突变的VAF值远低于1%时,常规NGS很难对真实的变异进行识别。
单细胞测序技术(single cell sequencing,SCS)可以在单一细胞水平实现精确的低频突变检测[37~39],包括单细胞测序[37],以及通过体外培养获得单细胞来源的细胞克隆[40]。如前所述,因为体细胞突变发生的丰度很低,并且对于正常组织每个细胞的突变都是唯一,而肿瘤组织也是异质性细胞克隆组成的混合体,经扩增后测序,突变与测序错误难以区分。克服这个问题的一种方法是使用基于单细胞的测序方法。SCS首先将单一的细胞从细胞群中分离出来,再进行全基因组扩增以获得足够的DNA用于文库制备及测序。进行突变分析时,以人类基因组为例,其基因组为二倍体;当发生体细胞突变时,其测序数据中对应位点的碱基信息将由纯合变为为杂合的,即~50%比对到参考基因组的序列信息为正常碱基,~50%为突变碱基信息。随着测序深度的增加,待测位点突变碱基的变异等位基因频率趋近于50%,远高于该位点NGS随机测序错误的发生率(~1%);因此,此时可以忽略随机测序错误的影响,得到可信度较高的突变信息。在一些研究中,利用SCS进行突变检测的错误率可以低至10–7~10–9水平[40]。然而,利用SCS进行突变检测仍然面临着诸多问题[41]。制备细胞悬液从中分离单个细胞是SCS的第一步。对于大量的实体组织而言,分离出具有良好扩增效率的单细胞仍存在一定的技术限制。此外,与常规测序方法不同,SCS是对单个细胞进行测序,因此很容易受到选择偏倚的影响。利用常规的机械法或酶法破碎组织、制备单细胞悬液时,会损失细胞在原始组织结构中的信息,例如发生组织病理学改变的区域与正常组织区域的细胞难以区分,导致在选择单细胞进行测序时产生偏倚,最终可能影响结果的分析。基因组扩增也是导致SCS突变分析产生偏倚的一个重要原因。由于SCS是从单个细胞进行扩增,加之PCR扩增反应固有的偏向性,很容易出现扩增产物的不均匀以及基因组覆盖度不足等问题,从而引起假阳性错误或者假阴性结果[39,42]。一些研究的结果显示,基因组扩增会产生较高水平(15%~64%)的假阴性结果[40]。另外,在实践中,为了降低SCS可能产生的偏倚,往往需要对多个单细胞进行测序,由此会带来相应的测序成本的升高。
针对单细胞测序的覆盖度低和错误率高等缺点,研究近年来也取得一定进展。单个细胞在全基因组进行扩增时容易引起核苷酸序列中产生错误,导致测序错误掩盖真实发生的突变。2017年,美国爱因斯坦医学院的Jan Vijg博士发表了一种能够准确鉴定单细胞基因组基因突变(单核苷酸变异分析)的新方法[37],该方法结合单细胞多重置换扩增(multiple displacement amplification,MDA)和单细胞变异鉴定(single-cell-variant caller,SCcaller),经验证可以修复基因扩增过程中发生的核苷酸序列错误、从而从单个细胞准确鉴定整个基因组的单核苷酸突变。该方法能够对几个单细胞进行测序,检测细胞中基因突变发生的频率,从而对患癌早期风险进行评估。此外,该方法还可以帮助揭示基因突变在人体衰老中的作用[43]。2023年,He等[44]发表了一项单细胞全外显基因组测序研究策略,将MDA与基因组多位点qPCR检测结合,通过单细胞分选、全基因组扩增、扩增均匀性评估和全外显子文库构建到测序分析,在外显子组捕获之前就排除有潜在扩增偏倚风险的单个细胞样本,显著提高了外显子组的覆盖率。该策略用于研究肝母细胞瘤肿瘤微环境,提升了肿瘤中的低频突变检出率。上述单细胞测序改进方案在检测人类疾病的低频突变、揭示疾病遗传异质性等方面的稳定性及重要性,为疾病演进的研究以及临床诊断和治疗提供有效的参考及指导。
2.1 校正测序错误的NGS技术(error-corrected next generation sequencing,ecNGS)
降低NGS测序错误率的策略主要包括建库实验条件优化、生物信息学分析方法改进和分析一致性测序。实验条件优化是针对测序过程中可能导致测序错误的实验步骤(DNA片段化过程和 PCR 扩增过程等),以减少文库制备和测序过程引入的错误,例如利用高保真酶进行 PCR 扩增以减少 PCR 扩增时引入的碱基错配、使用温和的文库制备条件或者加入 DNA 修复酶以减少文库制备过程带来的碱基损伤等[45];生物信息学分析方法改进包括对测序数据进行过滤,利用高质量的碱基/序列进行分析、优化序列比对算法,减少比对错误、根据突变碱基在读段的分布进行过滤,减少读段末端因比对错误导致的错配、要求突变出现在同一位点多条具有不同比对方向的独立读段上,减少单链损伤导致的测序错误以及利用平行对照样本进行系统误差的校正等[45]。然而,上述两种策略提高NGS突变检测准确性的能力仍然非常有限,仅能降低测序错误率至10–3左右。采用分子一致性测序策略(molecular consensus sequencing strategy)进行测序错误校正的分子一致性测序具有很高准确性,可实现在单个DNA分子水平上准确的突变检测[45],目前被认为是最有效的降低NGS测序错误率的策略。
该策略的基本原理是:利用同一模板来源的多个拷贝的测序信息(即多次测量)进行测序错误的校正,从而降低NGS的错误率(图2)。分子一致性测序通常使用PCR扩增来获得一条模板的多个拷贝片段,并利用模板上的分子标签(molecular barcodes)识别和标记这些从一条模板来源的拷贝片段的测序信息。随后,对这些拷贝片段分别进行测序并进行比对。理论上,这些同一模板来源的拷贝片段是同质的,其序列信息应当是完全一致的。同一位点上一致的碱基信息代表了该位点真实的碱基信息,其中少数不一致的变异信息则提示可能源于测序错误。通过这种比对,可以去除大量因随机测序错误导致的变异位点,得到与模板信息高度一致的序列信息用于突变检测。根据待检测的模板的不同,一致性测序分析又进一步可以分为基于单链的一致性测序方法(single strand consensus sequencing,SSCS)[46,47]和基于DNA互补双链的一致性测序方法(duplex consensus sequencing,DCS)[45]。DCS对DNA双链模板的两条单链同时进行测序错误的校正,能够进一步剔除在SSCS中不能去除的单链损伤所导致的测序错误,从而进一步提高了一致性测序分析的准确性,理论上可以将NGS碱基识别的错误率降低至10–9[48]。
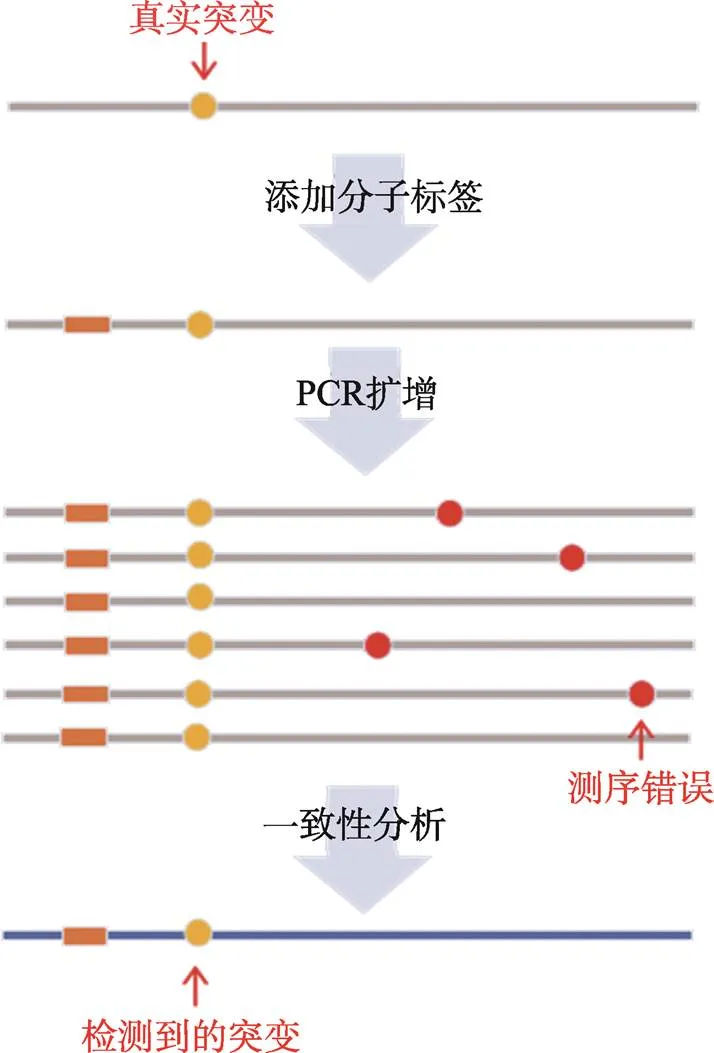
图2 分子一致性测序策略原理图
对待测模板添加外源性分子标签后进行PCR扩增及NGS测序,根据分子标签提取同一模板来源的PCR拷贝的序列。理论上,真实的变异(黄色圆点)会随着PCR扩增而在绝大多数拷贝中被观察到,而随机测序错误导致的变异(红色圆点)只会出现在部分拷贝中。利用这种区别,可以识别和去除由于随机测序错误导致的变异,从而提高NGS进行突变检测的准确性。当使用内源性标签时,无需另外添加分子标签,直接使用序列在基因组上的比对坐标作为分子标签提取同一模板来源的PCR拷贝序列。
2.2 分子一致性测序技术的研究进展
2012年,Schmitt等[48]首次报道了基于分子一致性策略的低频突变检测方法,即Duplex sequencing (DupSeq)。该方法引入了双链的单分子标签(unique molecular barcodes,UMIs)对DNA双链模板同时进行标记,从而将DNA双链的拷贝片段的信息结合起来进行测序错误校正,其可以将NGS的错误率降低至10–7以下[48,49]。DupSeq目前已经商业化,并被广泛用于对各种致突变剂和致癌物诱发突变的检测[34,50,51]。但是,DupSeq利用PCR扩增来获得大量目标基因片段的拷贝片段来进行测序错误校正,相对于常规的测序来说测序深度和测序成本很高,因此迄今为止主要用于对于少数目标基因片段或者较小的基因组范围进行检测。针对这一不足,Hoang等[52]采用Bottleneck sequencing system (BotSeqS)通过在进行PCR 扩增前对文库片段进行稀释的策略以及利用DNA片段的断裂位点作为内源性的UMIs,实现了对于人类基因组全基因组范围上的自发突变的检测。Matsumura等[53]采用与BotSeqS类似的策略,开发了Hypothesis alignment with weak overlap (Hawk-SeqTM),并应用于检测小鼠基因组上诱发突变的检测。作者团队也经过多年研究,利用了缩短的测序文库经双端测序产生的重叠片段,开发了一种不依赖于PCR扩增的DCS检测方法:DNA互补链双端测序一致性测序方法,命名为PECC-Seq (paired-end and complementary consensus sequencing)。技术原创点包括截短片段,PCR-free文库制备,DNA互补双链通过双端测序产生4条重叠片段,与参考基因组比对进行错误校正[54]。该策略降低了NGS测序的错误率,同时提高了测序效率从而降低了测序成本。后续又经深入分析,找到测序错误主要来源于文库制备过程中发生单链损伤,在末端修复过程中可能会向模版中引入难以去除的末端修复错误,从而干扰DCS检测的准确性,据此在文库制备时利用单链特异性的核酸酶对末端修复位点进行特异性切除,测序错误率进一步降低至~3.7×10–8[55]。Otsubo等[56]也报道了类似发现并采取相应优化措施,如使用单链特异性核酸酶的Jade-SeqTM(基于Hawk-SeqTM)。采用双脱氧核苷酸阻断损伤位点合成的NanoSeq (基于BotSeqS)由Sanger研究所开发,将NGS错误率进一步降低至~5×10–9,并且测序效率高,目前代表了本领域最前沿水平[57]。Bae等[58]提出Concatenating original duplex for error correction (CODEC)尝试了另一种思路,通过将DNA双链串联成单链进行DCS分析,可以同时兼容全基因组测序和靶向测序的策略。
此外,第三代测序平台的PacBio single-molecule real-time sequencing (SMRT)测序也可实现基于DCS的低频突变检测。三代测序边合成边测序,可提供目前最长的读取长度。将样本中的双链DNA通过两端加上接头构建哑铃状分子结构,得到圆环结构分子可进行滚环复制,通过对环状文库分子循环测序即可消除测序错误率[41]。基于上述原理的三代测序技术 High-fidelity sequencing (HiFi sequencing)已被证明可以在全基因组水平上检测细菌、线虫和哺乳动物细胞上的低频突变,测序准确性达到自发突变频率的水平[41]。利用同样策略加发夹结构接头、利用滚环复制扩增和错误校正的还有SMM-seq,该技术使用具有强链置换活性的人工耐热聚合酶对环状DNA单链分子进行有效且无偏倚放大后测序,应用在诱变剂和衰老研究中已实现了准确、经济高效的测序[59]。
分子一致性测序策略能够显著提高NGS用于低频突变检测的准确性,已应用于人群、小鼠和微生物等诸多场景的自发突变检测和致突变剂诱发突变的低频突变检测[50,52,53,57]。但是相较于常规的NGS检测,其所需的测序深度和测序成本很高。在常规的NGS测序中,理论上每一个模板至多只应当被检测一次以避免PCR重复片段带来的偏倚。而在分子一致性测序中,同一个模板来源的多个拷贝需要被测序以进行错误校正,因此分子一致性测序方法的测序深度比常规的NGS要高很多,由此带来测序成本昂贵的问题使得该技术的实际应用受限,现阶段大多报道仅用于靶向测序或用于小型基因组物种上的测序。因此,对于大型基因组(如人类和大小鼠基因组)全基因组范围的突变检测,如何降低分子一致性测序成本成为一个亟需解决的问题,例如BotSeqS和NanoSeq对文库片段依据算法进行稀释[52,57]、PECC-Seq利用PCR-free策略减少拷贝数[54,55],都是提高测序效率、降低测序成本的策略。在低频突变检测中,单一方法可能很难同时满足低频突变检测的广度和深度两个维度,需要根据具体研究目的进行方法的选择和设计。
3 低频突变NGS检测技术的应用展望
3.1 外源化学物的致突变性评价
长期以来,基于表型筛选的突变检测方法是最常用的评价外源化合物致突变性的手段[20~22],但是如前所述,均存在诸多局限性,低频突变NGS检测技术未来有望与一般毒性实验整合,替代经典体内致突变实验用于外源化学物致突变性的评价。将测序技术整合现有毒性检测终点,不仅能够用突变频率进行受试物致突变性的定性评价,还能通过定量分析推算安全阈值,最后还能进行突变特征分析更好地理解诱变机制。目前,已有关于ecNGS被用于细菌、哺乳动物细胞和小鼠模型中环境诱变剂诱发突变检测的研究报道[34,53,54]。作者团队在前期工作中,在delta转基因小鼠模型中同时应用PECC-Seq和基因突变实验对诱变剂马兜铃酸I诱导的突变进行检测,发现与基因突变实验相比,PECC-Seq在突变频率检测上表现出更高的灵敏度和特异性。此外,鉴定的化合物突变特征与临床马兜铃酸相关肿瘤上检测到的突变标签高度接近,而这样的突变谱是难以通过报告基因PCR产物测序(基因长度仅为456 bp)获得[55]。目前,国际多机构已经开展了对于ecNGS在致突变性和致癌性评价应用的讨论[60]。2023年,国际健康与环境科学研究机构(Health and Environmental Sciences Institute,HESI)下属的遗传毒性测试专家工作组开展了评估,随后,国际相关研究机构、多国制药企业和安评机构于2023年5月联合在杂志上发表评论文章[61],呼吁采取行动,进一步表征ecNGS并将其标准化,使其尽早成为被监管机构认可的重要遗传毒性测试方法列入指南,用于药物研发或环境诱变剂的风险评估。
3.2 细胞与基因编辑药物的风险评估
细胞与基因编辑作为革命性技术在全球范围内高速推进,在不同适应症领域内均展示巨大应用潜力,全球首款CRISPR基因编辑治疗药物Exa-cel在2023年底获批用于治疗镰状细胞性贫血病,成为领域的里程碑事件。与此同时,细胞与基因编辑治疗药物潜在的健康风险也愈发引起关注,近期FDA调查CAR-T治疗后有T细胞恶性肿瘤的严重风险,提示上述药物潜在的细胞恶性转化、基因编辑的插入突变和脱靶效应的风险亟需评估。但是,针对上述风险的筛查方案尚未达成普遍共识。目前多采纳的计算机预测工具(prediction,ISP)基于算法对sgRNA人类基因组同源性可能的错配序列进行比对和预测,结合靶向测序技术或全基因组测序考察可能的切割位点,以评估脱靶风险。但是由于现有数据库数据量有限、个体差异的存在和NGS高成本和高错误率,使监管机构对上述方案的检测能力存在担忧,而新兴的低错误率和低成本的ecNGS作为替代工具被期待,被国际领域专家联合呼吁尽早进行验证研究[61]。此外,针对基因修饰的免疫细胞或干细胞的成瘤性/致瘤性风险,目前体外细胞评价普遍采用的技术包括核型分析(评估遗传毒性)、软琼脂克隆形成实验(检测转化细胞)、端粒酶活性检测和数字PCR(检测未分化iPSCs或ESCs)。上述技术检测到的细胞多为表型已发生改变,而基因组上突变负荷的增加,或驱动基因已发生突变但是细胞尚未恶性转化的早期状态则无法被上述技术检测到。在体外培养阶段对细胞全基因组范围进行ecNGS,可考察基因突变发生频率是否升高,结合针对递送载体序列整合到宿主基因组中的预期位点进行靶向ecNGS,都有望在早期识别细胞致瘤性/成瘤性风险。
3.3 肿瘤发生发展的机制研究
肿瘤的发生是环境因素和遗传因素相互作用的结果。人类肿瘤的发生90%与环境因素有关,但真实世界中人类接触的环境物质极为复杂,阐明复杂环境因素与人类相关肿瘤的关联及机制是科学家持续努力解决的科学问题。不同的环境诱变因素会诱导各自不同的突变类型,包括碱基突变类型、突变发生的序列偏向性和突变发生的链偏向性等。科学家通过对大量肿瘤样本进行基因组测序和突变分析,得出的特征性印记被称为突变标签(mutational signature),突变标签对于了解肿瘤的发生机制、诊断和治疗具有重要意义。迄今已鉴定出超过50种的突变标签,例如吸烟可以引起以非转录链上G>T为主的突变(COSMIC数据库突变标签4);而中药致癌成分马兜铃酸暴露可以引起以T>A为主的颠换突变,并且易发生于非转录链的5′-CpApG-3′序列上(COSMIC数据库突变标签22)[1,62~64]。然而,上述突变标签是从肿瘤组织的突变数据中通过非负矩阵分解的统计方法分离的[65],其可能混杂了不同的致突变过程的作用、测序错误和数据分析引入的偏倚等,并不一定真实反映单一诱变因素的不良结局,仍有许多致突变过程和其突变特征之间尚未建立联系。将动物实验与人群流行病研究结果整合分析,有可能为阐明复杂环境因素与人类相关肿瘤发生的关联提供重要参考。借助基于NGS的低频突变检测手段,可以对实验动物给予单个诱变剂或混合物,在正常靶器官基因组上进行基因组的突变频率与突变特征分析。同时结合流行病学研究,在相关癌症患者上进行内暴露检测或DNA加合物组研究以分析致癌物暴露特征,利用NGS技术分析肿瘤组织基因组突变特征,与COSMIC数据库中的突变标签进行比对,同时把实验动物研究结果与临床研究数据进行关联分析,有可能鉴定出与肿瘤发生最为相关的环境因素。
低频突变检测技术对肿瘤的多阶段发展研究也具有潜在价值。不同阶段的突变特征动态地记录了机体经历的不同诱变过程,例如贯穿整个生命周期的胞嘧啶自发脱氨基作用、个体既往或者正在经历的外源性诱变剂的暴露和由于突变积累导致的DNA修复酶缺陷等[62,65]。同时,一些与疾病发生密切相关的关键基因的突变也提示了疾病发生发展过程中经历的关键步骤[66]。通过监测肿瘤发生发展过程中基因组上突变特征的动态变化以及关键基因的突变情况,将有可能揭示整个疾病发展过程中不同阶段不同致突变因素的作用情况,以及影响疾病发生进展的关键步骤,从而阐明肿瘤发生发展的机制以及可能的防治靶点。
3.4 人群肿瘤风险监测
生物学标志对于外源致癌物的暴露评估、致癌风险评估和癌症早期诊断和预防等方面具有重要作用,其主要包括效应标志、暴露标志和易感标志[67]。基因突变是肿瘤发生的起始事件,尤其是肿瘤相关基因上的驱动突变的不断积累意味着个体远期肿瘤发生风险的增加。目前,ecNGS检测低频突变的错误率可达10–7以下,接近人类基因组的自发突变水平[45,48,49,55,57,58],表明低频突变检测方法可以用于一般人群的体细胞的突变检测。基因组上突变频率的升高可以作为肿瘤早期效应标志用于预测患病风险。因此,对于暴露于特定环境致癌因素下的人群,例如高污染地区、职业暴露的健康高风险人群,或使用具有诱变作用药物治疗疾病的患者,在致癌因素相对已知的情况下,对其外周血或者尿液样本来源DNA,通过基于NGS的低频突变检测技术考察基因组上突变负荷的升高、与特定致癌物突变标签相关联的突变特征、疾病相关易感基因的突变状态等,将有助于监测其健康状态以及预测远期疾病风险。
除健康高风险人群外,低频突变检测技术还有望用于肿瘤患者的风险监测,因为低频突变与肿瘤的复发和耐药性的产生密切相关。微小残留病灶(minimal residual disease,MRD)是癌症患者在经过治疗后体内仍然存在难以被常规医学手段鉴别的残留肿瘤细胞,通过液体活检才能被发现[68]。治疗中可先对原发肿瘤测序筛查患者特异性突变的基因,术后对外周血中凋亡肿瘤细胞释放入血的DNA片段通过PCR技术或基于NGS的低频突变检测技术进行分析,评估肿瘤复发风险。MRD检测最早应用于血液肿瘤,目前用于实体瘤的临床实践研究也广泛开展。由于NGS成本的限制,很难实现全基因组或全外显子组超高深度测序,因此多利用靶向测序策略,例如CAPP-Seq通过筛选139个肺癌相关高频突变基因,缩小测序片段大小进而实现高准确性的超深度测序[69];Tam-Seq通过设计特异性引物对靶基因的目标区域进行PCR扩增之后再测序[70];Safer-Seq利用分子条形码以及巢式PCR区分正反义链,识别PCR及测序错误,进一步提高ctDNA突变检测的灵敏度及特异度[71]。近年来采用ecNGS技术的还有用于评估急性髓细胞性白血病MRD的报道,可以识别到VAFs在0.05%~0.10% 的相关突变,并提示了MRD状态与疾病复发和预后的相关性[72~74]。类似检测用于术后也有可能检测到治疗药物耐药性的发生,以此依据选择最佳用药方案。现阶段MRD检测仍然面临很多挑战,包括ctDNA丰度过低导致假阴性结果,肿瘤时空异质性导致所检测样本难以反映肿瘤整体情况,以及MRD的肿瘤细胞与原发肿瘤的突变发生变化等;另外,将低频突变检测技术用于人群的相关研究报道还很有限,还需要更多的人群研究结果挖掘其潜在应用价值。
3.5 生命科学研究及其他领域
以ecNGS为代表的低频突变检测技术的出现,为生命科学领域待解难题的体细胞突变、发育与衰老的深入研究提供了重要研究工具。2021年的一期杂志连续刊发四篇相关论文揭示人体体细胞突变规律。其中,利用NanoSeq技术的团队对人体不同组织进行突变分析,发现生殖细胞获得突变的速度异常的低,可能由于基底精原细胞的细胞分裂率较低,而结肠组织自发突发频率相对其他组织更高[75]。来自中国的研究团队则利用显微切割技术对肿瘤组织取样进行低频突变测序,发现不同正常组织器官的体细胞均存在大量的突变积累,而体细胞突变负荷及等位基因突变频率表现出明显的器官差异性,其中正常肝组织的体细胞突变负荷最高,而胰腺实质细胞中的突变负荷是最低的[76]。此外,在许多需要对高异质性样本检测的场景,例如对环境中耐药菌株的监测等,基于NGS的低频突变检测方法都可能成为新的检测和研究工具。
综上所述,经典的突变检测技术已得到广泛应用和验证,方法简便,可对外源化学物在早期突变筛查中进行快速检测,因此可针对不同目的与NGS技术进行互补使用。基于NGS的低频突变检测技术具有更高的准确性、更丰富的数据输出以及跨物种和跨位点的适用性,随着技术的不断发展,有望在药物研发领域、环境科学和生命科学基础研究领域不断扩大应用空间,为解明生命现象和活动规律、疾病准确诊断、降低环境健康风险以及保障临床用药安全等方面发挥更重要的作用,成为突变研究、监管安全测试和新兴临床应用的强大新工具。
感谢中国人民解放军海军军医大学的张天宝教授对文章给予的建议和讨论。
[1] Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jäger N, Jones DTW, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdés-Mas R, van Buuren MM, van 't Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome Initiative, ICGC Breast Cancer Consortium, ICGC MMML-Seq Consortium, ICGC PedBrain, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. Signatures of mutational processes in human cancer.,2013, 500(7463): 415–421.
[2] Haradhvala NJ, Polak P, Stojanov P, Covington KR, Shinbrot E, Hess JM, Rheinbay E, Kim J, Maruvka YE, Braunstein LZ, Kamburov A, Hanawalt PC, Wheeler DA, Koren A, Lawrence MS, Getz G. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair., 2016, 164(3): 538–549.
[3] Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution.,2013, 501(7467): 338–345.
[4] Beckman RA, Loeb LA. Evolutionary dynamics and significance of multiple subclonal mutations in cancer.,2017, 56: 7–15.
[5] Schmitt MW, Loeb LA, Salk JJ. The influence of subclonal resistance mutations on targeted cancer therapy.,2016, 13(6): 335–347.
[6] Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells., 2015, 349(6255): 1483–1489.
[7] Kucab JE, Zou XQ, Morganella S, Joel M, Nanda AS, Nagy E, Gomez C, Degasperi A, Harris R, Jackson SP, Arlt VM, Phillips DH, Nik-Zainal S. A compendium of mutational signatures of environmental agents., 2019, 177(4): 821–836.e16.
[8] Bielas JH, Loeb LA. Quantification of random genomic mutations., 2005, 2 (4): 285–290.
[9] Roach JC, Glusman G, Smit AFA, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, Shendure J, Drmanac R, Jorde LB, Hood L, Galas DJ. Analysis of genetic inheritance in a family quartet by whole-genome sequencing.,2010, 328(5978): 636–639.
[10] Milholland B, Dong X, Zhang L, Hao XX, Suh YS, Vijg J. Differences between germline and somatic mutation rates in humans and mice., 2017, 8: 15183.
[11] Besenbacher S, Liu SY, Izarzugaza JMG, Grove J, Belling K, Bork-Jensen J, Huang SJ, Als TD, Li ST, Yadav R, Rubio-García A, Lescai F, Demontis D, Rao JH, Ye WJ, Mailund T, Friborg RM, Pedersen CNS, Xu RQ, Sun JH, Liu H, Wang O, Cheng XF, Flores D, Rydza E, Rapacki K, Damm Sørensen J, Chmura P, Westergaard D, Dworzynski P, Sørensen TI, Lund O, Hansen T, Xu X, Li N, Bolund L, Pedersen O, Eiberg H, Krogh A, Børglum AD, Brunak S, Kristiansen K, Schierup MH, Wang J, Gupta R, Villesen P, Rasmussen S. Novel variation andmutation rates in population-wideassembled Danish trios.,2015, 6: 5969.
[12] Salk JJ, Kennedy SR. Next-generation genotoxicology: using modern sequencing technologies to assess somatic mutagenesis and cancer risk.,2020, 61(1): 135–151.
[13] White PA, Luijten M, Mishima M, Cox JA, Hanna JN, Maertens RM, Zwart EP.mammalian cell mutation assays based on transgenic reporters: a report of the international workshop on genotoxicity testing (IWGT).,2019, 847: 403039.
[14] Kirkland D, Uno Y, Luijten M, Beevers C, van Benthem J, Burlinson B, Dertinger S, Douglas GR, Hamada S, Horibata K, Lovell DP, Manjanatha M, Martus HJ, Mei N, Morita T, Ohyama W, Williams A.genotoxicity testing strategies: report from the 7th international workshop on genotoxicity testing (IWGT).,2019, 847: 403035.
[15] Gollapudi BB, Lynch AM, Heflich RH, Dertinger SD, Dobrovolsky VN, Froetschl R, Horibata K, Kenyon MO, Kimoto T, Lovell DP, Stankowski LF Jr, White PA, Witt KL, Tanir JY. ThePig-a assay: a report of the international workshop on genotoxicity testing (IWGT) workgroup.,2015, 783: 23–35.
[16] Nohmi T. Past, present and future directions ofdelta rodent gene mutation assays.,2016, 4(1): 1–13.
[17] Nohmi T, Masumura K, Toyoda-Hokaiwado N. Transgenic rat models for mutagenesis and carcinogenesis.,2017, 39: 11.
[18] Ames BN, Lee FD, Durston WE. An improved bacterial test system for the detection and classification of mutagens and carcinogens., 1973, 70(3): 782–786.
[19] OECD/OCDE. Test no. 470: mammalian erythrocyte Pig-a gene mutation assay, OECD guidelines for the testing of chemicals, section 4.,2022.
[20] OECD/OCDE. Test no. 471: bacterial reverse mutation test, OECD guidelines for the testing of chemicals, section 4.,2020.
[21] OECD/OCDE. Test no. 488: transgenic rodent somatic and germ cell gene mutation assays, OECD guidelines for the testing of chemicals, section 4.,2020.
[22] OECD/OCDE. Test no. 490:mammalian cell gene mutation tests using the thymidine kinase gene, OECD guidelines for the testing of chemicals, section 4.,2016.
[23] Nohmi T, Masumura K. Gpt delta transgenic mouse: a novel approach for molecular dissection of deletion mutations.,2004, 38: 97–121.
[24] Dertinger SD, Bhalli JA, Roberts DJ, Stankowski LF Jr, Gollapudi BB, Lovell DP, Recio L, Kimoto T, Miura D, Heflich RH. Recommendations for conducting the rodent erythrocyte Pig-a assay: a report from the HESI GTTC Pig-a workgroup.,2021, 62(3): 227–237.
[25] Parsons BL, Heflich RH. Genotypic selection methods for the direct analysis of point mutations.,1997, 387(2): 97–121.
[26] Postel M, Roosen A, Laurent-Puig P, Taly V, Wang-Renault SF. Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: a cancer diagnostic perspective., 2018, 18(1): 7–17.
[27] Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, Wyatt PW, Steenblock ER, Shah PH, Bousse LJ, Troup CB, Mellen JC, Wittmann DK, Erndt NG, Cauley TH, Koehler RT, So AP Dube S, Rose KA, Montesclaros L, Wang SL, Stumbo DP, Hodges SP, Romine S, Milanovich FP, White HE, Regan JF, Karlin-Neumann GA, Hindson CM, Saxonov S, Colston BW. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number.,2011, 83(22): 8604–8610.
[28] Chen LX, Liu PF, Evans TC Jr, Ettwiller LM. DNA damage is a pervasive cause of sequencing errors, directly confounding variant identification.,2017, 355(6326): 752–756.
[29] Costello M, Pugh TJ, Fennell TJ, Stewart C, Lichtenstein L, Meldrim JC, Fostel JL, Friedrich DC, Perrin D, Dionne D, Kim S, Gabriel SB, Lander ES, Fisher S, Getz G. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation.,2013, 41(6): e67.
[30] Do HD, Dobrovic A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization., 2015, 61(1): 64–71.
[31] Peng Q, Xu C, Kim D, Lewis M, DiCarlo J, Wang YX. Targeted single primer enrichment sequencing with single end duplex-UMI.,2019, 9(1): 4810.
[32] The Somatic Mutation Working Group of the SEQC-II Consortium. Achieving reproducibility and accuracy in cancer mutation detection with whole-genome and whole-exome sequencing.,2019, 626440.
[33] Maslov AY, Quispe-Tintaya W, Gorbacheva T, White RR, Vijg J. High-throughput sequencing in mutation detection: a new generation of genotoxicity tests?.,2015, 776: 136–143.
[34] Valentine CC 3rd, Young RR, Fielden MR, Kulkarni R, Williams LN, Li T, Minocherhomji S, Salk JJ. Direct quantification ofmutagenesis and carcinogenesis using duplex sequencing.,2020, 117(52): 33414–33425.
[35] Fox EJ, Reid-Bayliss KS, Emond MJ, Loeb LA. Accuracy of next generation sequencing platforms., 2014, 1: 1000106.
[36] Shendure J, Ji H. Next-generation DNA sequencing.,2008, 26(10): 1135–1145.
[37] Dong X, Zhang L, Milholland B, Lee M, Maslov AY, Wang T, Vijg J. Accurate identification of single- nucleotide variants in whole-genome-amplified single cells.,2017, 14(5): 491–493.
[38] Zong CH, Lu SJ, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell.,2012, 338(6114): 1622– 1626.
[39] Gonzalez-Pena V, Natarajan S, Xia YT, Klein D, Carter R, Pang YK, Shaner B, Annu K, Putnam D, Chen WA, Connelly J, Pruett-Miller S, Chen X, Easton J, Gawad C. Accurate genomic variant detection in single cells with primary template-directed amplification.,2021, 118(24): e2024176118.
[40] Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science.2016, 17 (3): 175–188.
[41] Revollo JR, Miranda JA, Dobrovolsky VN. PacBio sequencing detects genome-wide ultra-low-frequency substitution mutations resulting from exposure to chemical mutagens.,2021, 62(8): 438–445.
[42] Bai X, Li YX, Zeng XM, Zhao Q, Zhang ZW. Single-cell sequencing technology in tumor research.,2021, 518: 101–109.
[43] Zhang L, Lee M, Maslov AY, Montagna C, Vijg J, Dong X. Analyzing somatic mutations by single-cell whole-genome sequencing.,2023.
[44] He J, Meng M, Zhou XC, Gao R, Wang H. Isolation of single cells from human hepatoblastoma tissues for whole- exome sequencing.,2023, 4(1): 102052.
[45] Salk JJ, Schmitt MW, Loeb LA. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations.,2018, 19(5): 269– 285.
[46] Lou DI, Hussmann JA, McBee RM, Acevedo A, Andino R, Press WH, Sawyer SL. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing.,2013, 110(49): 19872–19877.
[47] Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing.,2011, 108(23): 9530–9535.
[48] Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. Detection of ultra-rare mutations by next-generation sequencing.,2012, 109(36): 14508–14513.
[49] Kennedy SR, Schmitt MW, Fox EJ, Kohrn BF, Salk JJ, Ahn EH, Prindle MJ, Kuong KJ, Shen JC, Risques RA, Loeb LA. Detecting ultralow-frequency mutations by duplex sequencing., 2014, 9(11): 2586–2606.
[50] Chawanthayatham S, Valentine CC 3rd, Fedeles BI, Fox EJ, Loeb LA, Levine SS, Slocum SL, Wogan GN, Croy RG, Essigmann JM. Mutational spectra of aflatoxin B1establish biomarkers of exposure for human hepatocellular carcinoma.,2017, 114(15): E3101–E3109.
[51] LeBlanc DPM, Meier M, Lo FY, Schmidt E, Valentine C 3rd, Williams A, Salk JJ, Yauk CL, Marchetti F. Duplex sequencing identifies genomic features that determine susceptibility to benzo(a)pyrene-inducedmutations.,2022, 23(1): 542.
[52] Hoang ML, Kinde I, Tomasetti C, McMahon KW, Rosenquist TA, Grollman AP, Kinzler KW, Vogelstein B, Papadopoulos N. Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing.,2016, 113(35): 9846–9851.
[53] Matsumura S, Sato H, Otsubo Y, Tasaki J, Ikeda N, Morita O. Genome-wide somatic mutation analysisHawk- SeqTMreveals mutation profiles associated with chemical mutagens.,2019, 93(9): 2689–2701.
[54] You XY, Thiruppathi S, Liu WY, Cao YY, Naito M, Furihata C, Honma M, Luan Y, Suzuki T. Detection of genome-wide low-frequency mutations with paired-end and complementary consensus sequencing (PECC-Seq) revealed end-repair-derived artifacts as residual errors.,2020, 94(10): 3475–3485.
[55] You XY, Cao YY, Suzuki T, Shao J, Zhu BZ, Masumura K, Xi J, Liu WY, Zhang XY, Luan Y. Genome-wide direct quantification ofmutagenesis using high-accuracy paired-end and complementary consensus sequencing.,2023, 51(21): e109.
[56] Otsubo Y, Matsumura S, Ikeda N, Yamane M. Single-strand specific nuclease enhances accuracy of error-corrected sequencing and improves rare mutation-detection sensitivity..2022, 96(1): 377–386.
[57] Abascal F, Harvey LMR, Mitchell E, Lawson ARJ, Lensing SV, Ellis P, Russell AJC, Alcantara RE, Baez-Ortega A, Wang YC, Kwa EJ, Lee-Six H, Cagan A, Coorens THH, Chapman MS, Olafsson S, Leonard S, Jones D, Machado HE, Davies M, Øbro NF, Mahubani KT, Allinson K, Gerstung M, Saeb-Parsy K, Kent DG, Laurenti E, Stratton MR, Rahbari R, Campbell PJ, Osborne RJ, Martincorena I. Somatic mutation landscapes at single-molecule resolution.,2021, 593(7859): 405–410.
[58] Bae JH, Liu RL, Roberts E, Nguyen E, Tabrizi S, Rhoades J, Blewett T, Xiong K, Gydush G, Shea D, An ZY, Patel S, Cheng J, Sridhar S, Liu MH, Lassen E, Skytte AB, Grońska-Pęski M, Shoag JE, Evrony GD, Parsons HA, Mayer EL, Makrigiorgos GM, Golub TR, Adalsteinsson VA. Single duplex DNA sequencing with CODEC detects mutations with high sensitivity.,2023, 55(5): 871–879.
[59] Maslov AY, Makhortov S, Sun SX, Heid J, Dong X, Lee M, Vijg J. Single-molecule, quantitative detection of low-abundance somatic mutations by high-throughput sequencing., 2022, 8(14): eabm3259.
[60] Lynch AM, Zanoni TB, Salk JJ, Martincorena I, Young RR, Kucab J, Valentine CC, Yauk C, Escobar PA, Witt KL, Frötschl R, Reed SH, Ashford A. Next generation sequencing workshop at the royal society of medicine (London, May 2022): how genomics is on the path to modernizing genetic toxicology.,2023, 38(4): 192–200.
[61] Marchetti F, Cardoso R, Chen CL, Douglas GR, Elloway J, Escobar PA, Harper T, Heflich RH, Kidd D, Lynch AM, Myers MB, Parsons BL, Salk JJ, Settivari RS, Smith-Roe SL, Witt KL, Yauk C, Young RR, Zhang SF, Minocherhomji S. Error-corrected next-generation sequencing to advance nonclinical genotoxicity and carcinogenicity testing.,2023, 22(3): 165–166.
[62] Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers.,2014, 15(9): 585–598.
[63] Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, Islam SMA, Lopez-Bigas N, Klimczak LJ, McPherson JR, Morganella S, Sabarinathan R, Wheeler DA, Mustonen V, PCAWG Mutational Signatures Working Group, Getz G, Rozen SG, Stratton MR, PCAWG Consortium. The repertoire of mutational signatures in human cancer.,2020, 578(7793): 94–101.
[64] Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, Campbell PJ, Vineis P, Phillips DH, Stratton MR. Mutational signatures associated with tobacco smoking in human cancer., 2016, 354(6312): 618–622.
[65] Nik-Zainal S, Kucab JE, Morganella S, Glodzik D, Alexandrov LB, Arlt VM, Weninger A, Hollstein M, Stratton MR, Phillips DH. The genome as a record of environmental exposure., 2015, 30(6): 763– 770.
[66] Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, Mularoni L, Pich O, Bonet J, Kranas H, Gonzalez-Perez A, Lopez-Bigas N. A compendium of mutational cancer driver genes., 2020, 20(10): 555–572.
[67] He YD. Genomic approach to biomarker identification and its recent applications.,2006, 2(3–4): 103–133.
[68] Yin JA, O'Brien MA, Hills RK, Daly SB, Wheatley K, Burnett AK. Minimal residual disease monitoring by quantitative RT-PCR in core binding factor AML allows risk stratification and predicts relapse: results of the United Kingdom MRC AML-15 trial., 2012, 120(14): 2826–2835.
[69] Gale D, Lawson ARJ, Howarth K, Madi M, Durham B, Smalley S, Calaway J, Blais S, Jones G, Clark J, Dimitrov P, Pugh M, Woodhouse S, Epstein M, Fernandez-Gonzalez A, Whale AS, Huggett JF, Foy CA, Jones GM, Raveh- Amit H, Schmitt K, Devonshire A, Green E, Forshew T, Plagnol V, Rosenfeld N. Development of a highly sensitive liquid biopsy platform to detect clinically- relevant cancer mutations at low allele fractions in cell-free DNA., 2018, 13(3): e0194630.
[70] Newman AM, Bratman SV, To J, Wynne JF, Eclov NCW, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, Shrager JB, Loo BW Jr, Alizadeh AA, Diehn M. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage.,2014, 20(5): 548–554.
[71] Cohen JD, Douville C, Dudley JC, Mog BJ, Popoli M, Ptak J, Dobbyn L, Silliman N, Schaefer J, Tie J, Gibbs P, Tomasetti C, Papadopoulos N, Kinzler KW, Vogelstein B. Detection of low-frequency DNA variants by targeted sequencing of the watson and crick strands., 2021, 39(10): 1220–1227.
[72] Patkar N, Kakirde C, Shaikh AF, Salve R, Bhanshe P, Chatterjee G, Rajpal S, Joshi S, Chaudhary S, Kodgule R, Ghoghale S, Deshpande N, Shetty D, Khizer SH, Jain H, Bagal B, Menon H, Khattry N, Sengar M, Tembhare P, Subramanian P, Gujral S. Clinical impact of panel-based error-corrected next generation sequencing versus flow cytometry to detect measurable residual disease (MRD) in acute myeloid leukemia (AML)., 2021, 35(5): 1392–1404.
[73] Hourigan CS, Dillon LW, Gui GG, Logan BR, Fei MW, Ghannam J, Li YS, Licon A, Alyea EP, Bashey A, Deeg HJ, Devine SM, Fernandez HF, Giralt S, Hamadani M, Howard A, Maziarz RT, Porter DL, Scott BL, Warlick ED, Pasquini MC, Horwitz ME. Impact of conditioning intensity of allogeneic transplantation for acute myeloid leukemia with genomic evidence of residual disease.,2020, 38(12): 1273–1283.
[74] Balagopal V, Hantel A, Kadri S, Steinhardt G, Zhen CJ, Kang WJ, Wanjari P, Ritterhouse LL, Stock W, Segal JP. Measurable residual disease monitoring for patients with acute myeloid leukemia following hematopoietic cell transplantation using error corrected hybrid capture next generation sequencing., 2019, 14(10): e0224097.
[75] Moore L, Cagan A, Coorens THH, Neville MDC, Sanghvi R, Sanders MA, Oliver TRW, Leongamornlert D, Ellis P, Noorani A, Mitchell TJ, Butler TM, Hooks Y, Warren AY, Jorgensen M, Dawson KJ, Menzies A, O'Neill L, Latimer C, Teng M, van Boxtel R, Iacobuzio-Donahue CA, Martincorena I, Heer R, Campbell PJ, Fitzgerald RC, Stratton MR, Rahbari R. The mutational landscape of human somatic and germline cells.,2021, 597(7876): 381–386.
[76] Li RY, Di L, Li J, Fan WY, Liu YC, Guo WJ, Liu WL, Liu L, Li Q, Chen LP, Chen YM, Miao CW, Liu HJ, Wang YQ, Ma YL, Xu DS, Lin DX, Huang YY, Wang JB, Bai F, Wu C. A body map of somatic mutagenesis in morphologically normal human tissues.2021, 597 (7876): 398– 403.
Application of next-generation sequencing in the detection of low-abundance mutations
Yang Luan1, Xinyue You1, Jin Yang2
Mutation accumulation in somatic cells contributes to cancer development, aging and many non-malignant diseases. The true mutation frequency in normal cells is extremely low, which presents a challenge in detecting these mutations at such low frequencies. The emergence of next-generation sequencing (NGS) technology enables direct detection of rare mutations across the entire genome of any species. This breakthrough overcomes numerous limitations of traditional mutation detection techniques that rely on specific detection models and sites. However, conventional NGS is limited in its application for detecting low-frequency mutations due to its high sequencing error rate. To address this challenge, high-accuracy NGS sequencing techniques based on molecular consensus sequencing strategies have been developed. These techniques have the ability to correct sequencing errors, resulting in error rates lower than 10–7, are expected to serve as effective tools for low-frequency mutation detection. Error-corrected NGS (ecNGS) techniques hold great potential in various areas, including safety evaluation and research on environmental mutagens, risk assessment of cell and gene therapy drugs, population health risk monitoring, and fundamental research in life sciences. This review highlights a comprehensive review of the research progress in low-frequency mutation detection techniques based on NGS, and provides a glimpse into their potential applications. It also offers an outlook on the potential applications of these techniques, thereby providing valuable insights for further development, research, and application of this technology in relevant fields.
next-generation sequencing; low-abundance mutations; molecular consensus sequencing; mutagenesis; risk assessment
2023-12-14;
2024-01-14;
2024-01-15
国家自然科学基金项目(编号:82304267)资助[Supported by the National Natural Science Foundation of China (No. 82304267)]
栾洋,博士,研究员,研究方向:遗传毒理学。E-mail: yluan@sjtu.edu.cn
10.16288/j.yczz.23-309
(责任编委: 卢大儒)
猜你喜欢
中国卫生统计(2023年5期)2023-11-30 01:40:14
英语世界(2023年6期)2023-06-30 06:29:10
今日农业(2021年11期)2021-08-13 08:53:24
中国生殖健康(2020年2期)2021-01-18 02:51:26
小学生导刊(2018年13期)2018-06-29 03:49:00
教师·中(2017年3期)2017-04-20 21:49:49
试题与研究·教学论坛(2016年27期)2016-08-11 14:57:08
教学研究与管理(2014年4期)2014-05-16 22:44:12
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
