
基于动态图卷积与迁移学习的蛋白质质量评估
2024-02-21 06:00冯子健黄伟鸿姜博文
软件导刊 2024年1期
冯子健,黄伟鸿,姜博文
(浙江理工大学 信息科学与工程学院,浙江 杭州 310018)
0 引言
蛋白质是一种由多个氨基酸分子组成的链,它是生物界的重要组成物质,参与了大多数生命活动。预测、理解和分析蛋白质对医学、遗传学、药学等领域具有重要意义[1]。随着新一代测序技术的发展,研究者能很容易获得蛋白质的氨基酸序列,然而蛋白质的功能却主要由其三维结构决定。
传统生化实验方法获取蛋白质结构既昂贵又耗时,为了能快速、高效获得蛋白质三维结构,计算方法便成为了一种重要的补充手段,在近年来得到了快速发展。然而,目前仍未存在一种方法的预测结果能完全达到生化实验的准确性,只能在特定环境下达到理想效果,因此评估计算方法便输出的蛋白质模型成为了实验中不可缺少的一环。
质量评估(Quality Assessment,QA)的目的就是将预测的蛋白质模型与天然结构进行比较,进而输出预测模型在各种评价指标上的得分,便于后期筛选出更接近天然结构的优秀模型。
AlphaFold2[2]在CASP14 中的表现标志着深度学习正颠覆性地改变蛋白质结构预测领域,但其在质量评估领域的应用还有待开发。为此,本文提出一种基于动态图卷积神经网络[3](Dynamic Graph CNN,DGCNN)的蛋白质模型质量评估方法DGCQA,并结合迁移学习思想,选用预训练模型ESM-1b[4]的编码特征作为输入。在CASP13[5]数据集上的实验表明,所提模型相较于同类方法效果更好。
1 相关工作
蛋白质模型质量评估方法大致可分为共识方法和单模型方法。共识方法通过一个候选池中的其他模型信息来评估蛋白质模型,它是质量评估发展初期的主流方法之一,尽管共识方法可在一些情况下取得较好的效果,但很大程度上受候选池中模型的影响,如果模型较少或缺少一致性和相似性时,将难以对蛋白质模型作出最正确的评价。单模型方法是将单个蛋白质结构作为输入,首先提取结构中的特征信息,然后通过机器学习、深度学习等方法进行训练。
从CASP13 开始,单一模型方法在质量评估领域开始有逐渐超越共识方法的趋势。ProQ2[6]、ProQ3[7]是质量评估中机器学习的典型方法,使用人工微调的特征来训练支持向量机(Support Vector Machine,SVM),但由于机器学习的局限性,该类方法难以学习蛋白质结构中更为复杂的特征关系。
近年来,伴随着深度学习的快速发展,一些基于深度学习的方法开始逐渐涌现。Hou 等[8]采用多任务学习训练了一个1DCNN 网络来评估蛋白质的全局和局部质量,但蛋白质存在多个残基,难以捕获残基间的长距离依赖关系。DeepAccNet[9]通过结合2D 和3D 卷积来预测每个残基的准确度,其中3D 卷积用于评估当前蛋白质的局部原子环境,2D 卷积则提供全局上下文信息。Nie 等[10]提出一种基于多尺度卷积和双向门控循环神经网络的质量评估方法BMBQA,结合数据增强思想,在原数据集中加入大量同源蛋白质来提升模型性能,但数据增强不仅增加了训练负担,还可能引入额外噪声。
图数据结构能方便、直观地表示蛋白质中的原子、氨基酸及他们之间的相互作用关系。ProteinGCN[11]、GraphQA[12]将蛋白质模型表示为图形式,通过图卷积神经网络(Graph Convolutional Network,GCN)[13]提取蛋白中残基的特征,再利用这些特征评估蛋白质模型质量。虽然,该方法效果较好,但图神经网络在使用前需要构建蛋白质的图结构,而图中的边往往需要根据残基间的距离进行构建,如果构建边的阈值选取不合理或蛋白质模型中的数据不够准确,将造成图中产生错误的边或边缺失等问题,会在一定程度上影响GCN 的效果。
为此,本文总结前人工作经验,针对图网络在蛋白质质量评估中存在的问题,提出基于动态图卷积神经网络的单一模型质量评估方法,并结合预训练模型ESM-1b 编码特征进一步提升模型效果。
2 实验方法
2.1 输入特征
本文输入为多种特征组合,可将其划分为残基特征和残基之间的成对特征。为了方便表述,设输入的蛋白质中残基个数为L,ri表示蛋白质序列中索引为i的残基,具体的残基特征如下。
2.1.1 one-hot编码
one-hot 编码用来表示蛋白质中残基类型的二进制向量,其中一位为1,其余位置为0。本文工作中,每一个残基被编码为一个长度为21 的向量(包括20 种标准氨基酸和其他氨基酸),蛋白质one-hot编码大小为L× 21。
2.1.2 残基相对位置编码
该特征向量用来表示每个残基在它所在蛋白质序列中的相对位置,计算公式如式(1)所示。
2.1.3 三维坐标
本文从PDB 文件中提取所有原子的三维坐标(x,y,z),并用Cβ原子的坐标代表残基坐标。
2.1.4 统计量
本文参考Hurtado 等[14]的工作,从蛋白质多序列比对(Multiple Sequence Alignment,MSA[15])中提取自信息量(self-information)和部分熵(partial entropy),如式(2)、式(3)所示。
式中:pi表示残基ri出现在当前位置的频率表示数据集的平均频率,两种统计量均为23维向量。
2.1.5 DSSP特征
DSSP 特征为DSSP[16]计算得到的二面角(dihedral angle)、相对可溶性(relative solvent accessibility)和二级结构(secondary structure)类型,并他们拼接为一个6维向量。
2.1.6 ESM-1b编码
本文受到迁移学习思想启发,将Facebook AI Research提出的蛋白质预训练模型ESM-1b 作为固定的特征编码器,将蛋白质序列编码为一个L× 1 280 的向量,并在输入前通过一维卷积降维到32 维,与其他特征拼接作为最终输入。ESM-1b 是一个基于Transformer[17]的无监督预训练模型,在具有上亿个蛋白质序列的数据库UniProt[18]上进行训练,可挖掘蛋白质序列中的结构信息。
综上,残基对特征的残基间序列间隔表示蛋白质序列中的两两残基间的索引距离(即两残基的索引差),用onehot 编码表示距离间隔[1,2,3,4,5,6-10,11-15,16-20,>20],大小为L×L× 9。残基间方位包括由trRosetta[19]定义的ω、θ二面角及φ平面角。
2.2 基于动态图卷积的蛋白质模型质量评估方法
2.2.1 Inception模块
本文选用Szegedy 等[20]提出的Inception 结构提取残基对特征,模型结果如图1 所示。由此可见,该结构为多尺度卷积核组成的网络模块,模块中3×3 和5×5 卷积核可让网络捕获不同大小邻域内的残基对信息,为下游任务提供更丰富的特征表达。此外,为了减少网络参数、提升训练速度,首先使用1×1 卷积核降维特征,后续进行大尺度卷积操作。

Fig.1 Incepiton module图1 Inception模块
2.2.2 EdgeConv层
GCN 等图神经网络在输入前就已构建了邻接矩阵,节点与节点间的邻接关系固定不变。以蛋白质为例,一个蛋白质图G一般通过以下方法构建:
式中:e为图的边;v为图的节点;i、j为残基索引;C代表残基中的Cβ原子坐标;dmax为构建边的阈值。
当前,尽管GCN 在质量评估领域取得了优异成果,但随着网络层次加深,残基间的邻接关系逐渐从初始的欧几里得距离变成高维的特征间距离,初始的邻接矩阵不再适合表达这种深层联系。因此,根据特征构建动态的邻接关系尤为重要。
EdgeConv 原本是一种面向点云学习的网络模块,本文将残基类比为特征空间上的点云,应用该模块挖掘残基的特征信息。设R={r1,r2…rn}⊆RF为蛋白质的残基云,任意一个残基表示为ri,邻域节点表示为rj,F为残基特征的通道数。如图2(a)所示,残基ri在EdgeConv 层中计算与其他所有残基的特征距离|ri-rj|,选取距离最小的k个残基作为其在本层的邻域(图中k=4),整个过程动态变化,即每一个EdgeConv 层都会重新计算ri的邻域来适应特征的变化。图2(b)展示了动态图网络的节点更新过程,计算公式如式(5)所示。
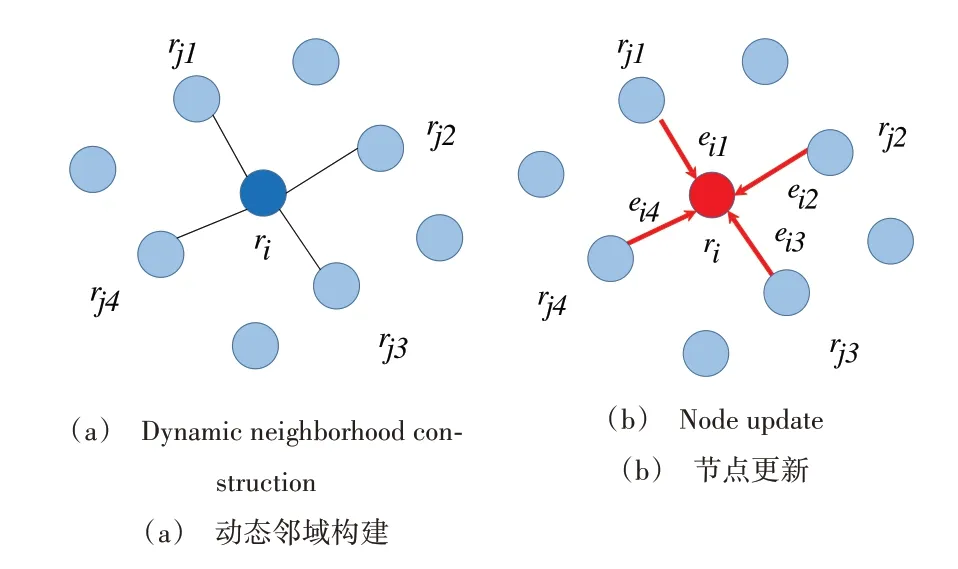
Fig.2 EdgeConv operation图2 EdgeConv操作
式中:ε表示ri与邻域节点rj构成的边集合。
hθ的定义为:
式中:为MLP,操作对象为ri的全局特征(ri)和局部边特征(ri-rj)。
2.2.3 总体架构
本文基于Inception、EdgeConv 模块,搭建了网络整体架构DGCQA,如图3 所示。网络初始输入的节点并不是残基,而是原子,原子特征选用其三维坐标(x,y,z)。DGCQA 首先在原子尺度上进行两次EdgeConv 操作(邻域范围k=40),以充分挖掘残基的原子几何特征,再通过Cβ原子特征代替残基,降采样到残基尺度的点云后进行后续3 层的EdgeConv 操作(邻域范围k=10)。

Fig.3 DGCQA architecture图3 DGCQA结构
网络中,同尺度下不同EdgeConv 层输出都会进行拼接,以充分利用网络在不同深度下的特征信息。最后,模型将特征压缩至适应大小,并与多尺度卷积模块的输出拼接,通过全连接层和sigmoid 函数(图3 中的σ)得到预测结果。EdgeConv 层的网络设计如图4 所示,首先获取残基节点k邻域范围内的节点信息,然后使用MLP 对所有节点及其邻域特征进行处理,最后通过池化层聚合特征,Edge-Conv层前后节点数量保持一致。
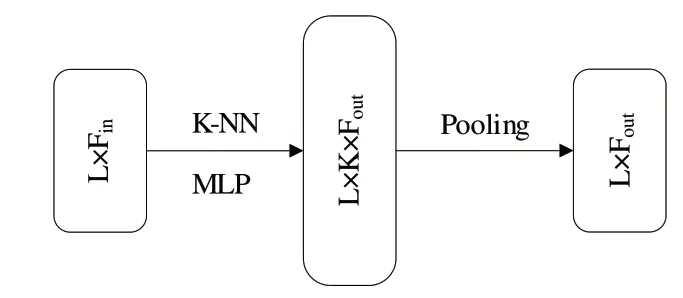
Fig.4 EdgeConv layer图4 EdgeConv层
DGCQA 结构中的多尺度卷积模块如图5 所示,该模块用于捕获不同视角下的残基对特征关系。以残基对特征作为输入,经过3 层Inception 结构的处理后通过pooling 层进行降采样,然后将特征展平为输出通道数Mout大小。
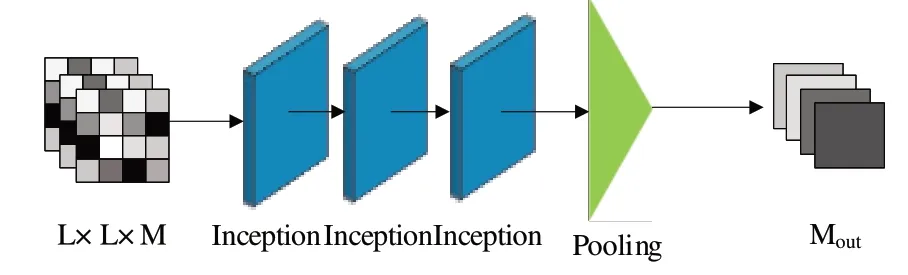
Fig.5 Multi-scale convolution module图5 多尺度卷积模块
3 实验结果及分析
3.1 实验环境
本文实验基于Ubuntu 18.04 操作系统,Pytorch 1.13.0深度学习框架,处理器为AMD Ryzen 95900X,显卡为NVIDIA RTX3090。
3.2 数据集
实验数据集来自CASP 9~CASP 13 比赛中官方提供的目标蛋白质和各参赛小组提供的预测蛋白质。针对每一个目标蛋白质(target),均有多个小组提交的候选模型(decoy)与之对应。本文将CASP 9~CASP 12 整合成85 000 个蛋白质的数据集,以目标蛋白质为基准随机划分训练集(270 个目标蛋白质)和验证集(50 个目标蛋白质)。同时,将CASP 13 阶段两个比赛中的14 000 个蛋白质模型(对应72 个目标蛋白质)作为测试集,将本文所提方法与其他方法进行比较。
3.3 评价指标
本文选用皮尔森相关系数(Pearson Correlation Coefficient)、斯皮尔曼相关系数(Spearman Correlation Coefficient)、均方根误差(Root Mean Square Error)、均方根误差(Root Mean Square Error)、标准分数(standard score)分析DGCQA 的性能,如式(7)—式(10)所示。
皮尔森相关系数用于评价两个向量之间的线性关系,取值范围为[-1,1],接近0 表示弱相关,接近-1 或1 表示强相关。
式中:Y代表真实值向量;代表预测值向量;M代表对应向量的平均值。
斯皮尔曼相关系数用于统计两个连续变量之间的单调关系,对异常值敏感度较低,其值与变量具体值无关,仅与变量间的大小关系有关。
式中:R为预测值的取值等级;S为真实值的取值等级;MR、MS分别为R、S的均值。
均方根误差是回归模型的典型评价指标,用于衡量预测值与真实值之间的偏差。
式中:n为预测的实例数量;yi为真实值为预测值。
标准分数也叫z 分数(z-score),本文在评估全局质量分数GDT_TS[21](Global Distance Test_Total Score)时,根据DGCQA 的预测结果,从每个目标蛋白质的候选模型中选择一个预测评分最高的蛋白质模型,并计算z-score 的平均值,值越高代表DGCQA 对优秀蛋白质模型的选择越准确。
式中:X为原始数据;为数据集的均值;σ为数据集的标准差。
3.4 结果分析
3.4.1 整体性能
由于蛋白质的全局结构精度分数GDT_TS 是CASP 评价蛋白质模型质量的重要指标,因此本文在全部蛋白质目标上计算GDT_TS 的均方根误差(RMSE)、皮尔森相关系数(R)、斯皮尔曼相关系数(ρ)和标准分数(z),并与12 种方法进行比较,具体数据如表1所示。

Table 1 GDT_ TS prediction evaluation indicators表1 GDT_TS预测评价指标
表1 中Rtarget为先在每个目标蛋白质的候选模型中计算皮尔森相关系数,然后再求均值,值越高表明该方法能更好地根据具体目标蛋白质候选模型的整体质量进行排名。由此可见,本文所提方法虽然在标准分数z 上相较于GraphQA 较差,但也具有一定的竞争力,相较于其他方法在多项指标达到最优。图6 显示了GDT_TS 的真实值与预测值的关系及其分布情况。

Fig.6 GDT_ TS marginal图6 GDT_TS边际
3.4.2 邻域范围影响
本文中动态图卷积的邻域范围是通过模型中Edge-Conv 层的超参数k所设置,k的取值会在一定程度上影响模型性能。实验中保持DGCQA 整体架构不变,仅修改在残基尺度上提取特征时的k邻域大小,观察模型在皮尔森系数下的得分情况。如图7 所示,当邻域范围k=10 时,模型的皮尔森相关系数达到最优效果0.876,而当邻域选取变小或变大时性能呈下降趋势。原因为当邻域选取过小时会导致动态图卷积视野受限,无法充分捕获节点间的特征关系;当邻域选取过大会使深层网络下的节点特征过于相似,将影响模型对不同节点的区分能力。

Fig.7 Pearson correlation coefficient under different neighborhood ranges图7 不同邻域范围下的皮尔森相关系数
3.4.3 迁移学习有效性分析
为了验证迁移学习的有效性,保持模型框架和其他特征输入不变,仅去除ESM-1b 编码以评估该部分对模型的影响。如表2、图8 所示,加入ESM-1b 特征能使DGCQA 在所有指标上存在不同程度的提升,尤其在Rtarget上ESM-1b特征帮助DGCQA 提升了0.032,超过了第二名GraphQA 的0.779,达到了目前最好效果。

Table 2 Comparison of predictions for the presence and absence of ESM-1b on the test set表2 测试集上有无ESM-1b的预测比较

Fig.8 ESM-1b feature impact图8 ESM-1b特征影响
3.4.4 动态图卷积效果分析
为了通过实验公平比较传统图卷积和动态图卷积在蛋白质模型质量评估上的性能。在动态图卷积方法上选择了仅使用EdgeConv 层搭建的简单网络DGCQA-RAW;在特征上使用蛋白质的氨基酸one-hot 编码作为GCN 和动态图卷积神经网络的唯一输入。
二者在5 个重要指标的得分如表3 所示。由此可知,DGCQA-RAW 的5 个指标中4 个超过GCN,证明在质量评估任务中,图节点的动态邻域相较于固定邻接关系对特征发掘更有效。

Table 3 Comparison of GCN and DGCNN performance in quality assessment表3 GCN和DGCNN在质量评估中的性能比较
4 结语
本文提出基于动态图卷积的质量评估方法DGCQA 来解决传统图卷积的局限性,并结合迁移学习思想,引入蛋白质预训练模型ESM-1b 编码特征训练模型。实验表明,DGCQA 的表现相较于传统方法更优秀,证明了所提方法的有效性。
然而,本文方法仍存在一定的缺陷。例如,动态图虽然无需构建固定邻接关系,但构建节点邻域范围的选择依然是一个不确定因素,选取的大小会在一定程度上影响模型性能。下一步,将构建一个对邻域范围具有自适应能力的动态图神经网络,以提升模型的鲁棒性。
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
老年教育(2021年11期)2021-12-12
老年教育(2021年10期)2021-11-10
老年教育(2021年8期)2021-08-21
老年教育(2021年3期)2021-03-22
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
吉林大学学报(理学版)(2020年3期)2020-05-29
自动化学报(2018年7期)2018-08-20
周口师范学院学报(2016年5期)2016-10-17
池州学院学报(2015年3期)2016-01-05
