
生物酶技术在二氧化碳转化中的应用研究进展
2024-02-20 06:50高子馨
化工环保 2024年1期
刘 恋,王 新,高子馨,鲍 佳
(沈阳工业大学 环境与化学工程学院,辽宁 沈阳 110870)
由于温室气体的大量排放,全球变暖已成为地球面临的严峻挑战。二氧化碳(CO2)的不节制排放是导致全球变暖的主要原因。减少CO2排放、开发低耗高效的CO2资源化利用新技术已成为当务之急[1]。基于在CO2转化中的独特优势,生物酶技术逐渐引起了人们的关注。在已发现的自然界6条天然固碳途径中,大部分是由核心固碳酶催化完成固碳。但这些核心固碳酶催化速率慢,反应过程复杂,且难以对其进行改造和优化。构建新型生物固碳酶,克服传统固碳酶的不足,提升其催化活性,已成为合成生物学领域的一个重要课题[2]。另外,在CO2的循环利用领域,生物酶催化因其高效、高选择性、条件温和等优势而备受青睐。碳酸酐酶(CA)和甲酸脱氢酶(FDH)是催化过程中关键的两种酶,前者能极大地促进CO2的水合,后者能将CO2还原为甲酸,二者协同能提高CO2的还原速率。但在实际生产中,由于温度、pH等原因,酶易失去活性[3],故需对其稳定性进行深入研究。通过对生物酶进行固定化,可提高其稳定性。
本文介绍了天然及人工固碳途径中核心固碳酶的改造与设计,总结了CA和FDH的固定化及其在CO2资源化转化中的应用研究进展,以期为其工业化应用提供参考。
1 生物酶在固碳途径中的改造与设计
1.1 天然固碳途径中固碳酶的改造与表达
卡尔文循环是最主要的天然固碳途径,其核心是核酮糖-1,5-双磷酸羧化酶/加氧酶(Rubisco)。近年来,大量研究对卡尔文循环进行了改造优化,其中的大多数集中在对Rubisco的改造优化上。CAI等[4]开发了一种基于大肠杆菌的活性导向选择系统,该系统将宿主细胞的生长仅与其中的Rubisco的活性相关联,通过一轮进化得到的PCC7002 Rubisco突变体,其比羧化活性提高了85%,对CO2的催化效率提高了45%。GLEIZER等[5]设计并进化了大肠杆菌,可将CO2转化为所有生物质碳,利用Rubisco和磷酸核酮糖激酶与FDH共表达,通过卡尔文循环实现了CO2的固定和还原。
乙酰辅酶A羧化酶(ACC)和丙酰辅酶羧化酶(PCC)是3-羟基丙酸双循环的核心固碳酶,可通过进化改造提高其固碳性能。LIU等[6]在重组大肠杆菌中表达了7种来自不同微生物的PCCB基因编码CT亚基,来自枯草芽孢杆菌的PCCB表现出最高的体外活性,通过定向进化进一步提升其活性,得到的新型PCC的总催化效率提高了94倍。
二羧酸/4-羟基丁酸循环的核心固碳酶磷酸烯醇丙酮酸羧化酶(PEPC)活性高且结构简单,故相关研究主要是以PEPC为核心固碳酶设计人工固碳途径。BAR-EVEN等[7]使用PEPC,构建了新型替代碳固定途径—丙二酰辅酶A-草酰乙酸-乙醛酸(MOG)途径,与卡尔文循环相比,MOG途径的通路特异性活性高出2~3倍。BOUZON等[8]利用PEPC将CO2转化为甲醛,然后将其转移至四氢叶酸中,进行后续反应。YU等[9]利用PEPC设计了苹果酰辅酶A-甘油酸途径(MCG),以补充卡尔文循环的不足,从而有效合成乙酰辅酶A,该途径在光合生物聚球藻中实施后,使碳酸氢盐同化速率提高了约2倍。
1.2 利用高效固碳酶的人工生物固碳
除了天然固碳途径中的固碳酶外,自然界中仍有许多活性很高的固碳酶。以这些固碳酶为基础设计全新固碳途径以实现人工生物固碳,是提高固碳效率的有效途径。
XIAO等[10]利用丙酮酸合酶设计了仅包含4个反应的最小化人工固碳循环—POAP循环。该POAP循环可在每个步骤中将两分子CO2转化为一分子草酸盐,代价是两分子ATP(三磷酸腺苷)和一还原当量(NAD(P)H(还原型辅酶Ⅰ/Ⅱ)形式)。该POAP循环可在50 ℃的厌氧条件下运行,固碳速率达到8.0 nmol/(min·mg)(以固碳酶计)。SIEGEL等[11]通过计算设计获得了一种甲醛酶(FLS),FLS催化碳化反应可直接将一碳单元固定成三碳单元。利用FLS设计了一种新的固碳途径,即甲醛酶途径,该途径比任何天然单碳同化途径利用碳更有效,并且具有更少的反向通量。SCHWANDER等[12]设计了一种在体外连续固定CO2的人工循环—巴豆酰辅酶A/乙基丙二酰辅酶A/羟基丁酰辅酶A(CETCH)循环。CETCH循环是一个由17种酶组成的反应体系,以5 nmol/(min·mg)(以核心固碳酶计)的速率将CO2转化为有机分子,比自然生物系统的效率更高,为6种天然固碳途径增加了人工替代途径。2021年,人工生物固碳领域又迎来了重大突破。CAI等[13]报道了一种在无细胞系统中由CO2和H2合成淀粉的化学-生物化学混合途径—人工淀粉合成代谢途径(ASAP)。该途径由11个核心反应组成,在时空隔离的化学酶系统中,在氢气驱动下,以22.0 nmol/(min·mg)(以总催化剂计)的速率将CO2转化为淀粉,较玉米中淀粉合成速率高约8.5倍。该途径为未来利用CO2合成生化杂交淀粉提供了思路。
1.3 小结
目前,对CO2固定途径的研究逐渐从天然固碳途径的改造与优化,过渡为人工固碳途径的全新设计、构建与应用。研究人员通过强化CO2羧化途径、优化CO2固定能量供给来提高CO2固定效率,通过重构CO2固定途径、开发设计全新的人工固碳途径来更好地实现CO2的资源化利用[14]。全新人工固碳途径与传统卡尔文循环的比较见表1。

表1 全新人工固碳途径与传统卡尔文循环的比较
2 CA催化CO2吸收
CA是一种锌基金属酶,通常存在于自然界的哺乳动物、植物、藻类、古生菌、脊椎动物和细菌中。它调节着人类和其他生物体的生物过程,其最主要的功能是催化CO2的可逆水合反应。单纯的CO2水合过程十分缓慢,一级反应速率常数仅为5×10-2s-1,但CA将CO2转化为碳酸氢盐的速率非常快,CA周转率为每分子CA每秒104~106分子CO2,是捕获封存CO2的最优酶[15]。
但CA酶对工作条件要求较高,且难以回收与再循环,这限制了其大规模工业应用。此外,使用游离酶会使酶总成本变得很高,酶变性会导致酶水化活性随着时间推移而逐渐丧失。酶固定化可以解决这些问题,使反应器设计更加灵活,同时提高了酶的回收率、稳定性和可重复使用性,降低了酶的总成本。
2.1 CA固定化载体材料
为了有效地重复使用酶,并降低封存过程成本,研究人员尝试在各种材料上固定CA。载体材料对生物催化体系的性能有重要影响,选择时应考虑成本、功能基团可用性、机械稳定性、生物降解性等特性[16]。
在酶的固定化方面,二氧化硅(SiO2)是应用最广泛的无机载体材料之一。硅基材料的使用形式多样,如溶胶-凝胶SiO2、SiO2纳米颗粒等[17],其大比表面积和多孔结构可以很好地结合酶。SHAO等[18]采用化学成分相同但物理结构不同的SiO2基介孔分子筛KIT-6、SBA-15和MCM-41作为CA的固定化载体。与游离酶相比,CA/KIT-6、CA/SBA-15和CA/MCM-41的半衰期分别延长了3.0、2.8和2.0倍。SBA-15因其二维结构和大孔径而表现出更高的CA负载量,在40 ℃下6 d后仍保持96%的初始活性,使CA/SBA-15较CA/MCM-41和CA/KIT-6更适合于CO2捕获。
当使用固定化酶时,催化过程完成后分离生物催化剂是新的挑战。将酶分子附着在磁颗粒上是解决方案之一。金属氧化物纳米颗粒具有大比表面积、高生物相容性以及与胺/羧基的配位能力,被广泛用于CA的固定化研究。VINOBA等[19]利用戊二醛作为间隔剂,探索了将牛的CA固定在包膜磁性纳米颗粒基质上的方法,在Fe3O4纳米颗粒上涂覆SiO2和OAPS(八氨基苯基倍半硅氧烷)。固定化CA在30次循环后表现出良好的可重复使用性,30 d后仍保持82%的初始活性,表明其具有CO2封存应用的潜力。
使用合成聚合物作为载体材料,其优势是可以选择不同的聚合单体来满足酶和固定化过程的需要。聚合物结构中可以观察到非常广泛的化学官能团,如羰基、羧基、强疏水的烷基等。这些基团有助于聚合物表面的功能化以及与酶的高效结合。对于酶的固定化,通过共价交联将CA附着在聚氨酯泡沫上,可提高CA的热稳定性,在低于50 ℃时可保持98%以上的活性[20]。
含有大量附着位点的多孔高吸水性水凝胶亦是理想的固定化载体。WEN等[21]将合成的CA双金属杂化纳米花(CANF)包埋于聚乙烯醇(PVA)-壳聚糖(CS)水凝胶网络中,制备了PVA/CS@CANF水凝胶膜,其在热稳定性、pH稳定性、可重复使用性、CO2捕获能力等方面均优于游离CA和CANF。JUN等[22]以酶沉淀涂层(EPC)的形式将CA固定在电纺聚合物纳米纤维上,室温下在水溶液中以200 r/min振荡培养868 d后,固定化CA仍保持65.3%的初始活性。
金属有机骨架(MOFs)是由金属离子和有机配体排列组装而成的独特的大比表面积多孔晶体材料。MOFs在固定化后的酶活性保留方面具有潜在优势。LIANG等[23]提出了在MOFs中原位嵌入蛋白质的想法。ASADI等[24]以微孔咪唑沸石骨架ZIF-8固定CA。对于Ni基MOFs,由于Ni-BTC的特殊结合能力,重组的人碳酸酐酶Ⅱ(hCA Ⅱ)可以很轻松地固定在Ni-BTC纳米棒上,在最佳条件下,来自细胞裂解物的组氨酸标记的hCA Ⅱ(His-hCAⅡ)可获得99%的活性回收率;储存10 d后,固定化His-hCA Ⅱ保留了40%的活性,而游离酶失去了91%的活性[25]。
2.2 固定化CA催化CO2捕获技术
气液填充床柱是研究较多的CO2捕获技术,填料提供大的表面积,以确保气液相之间的良好接触。在酶促CO2捕获情况下,填料可用作固定化支撑。BLAIS等[26]提出了一种将CA共价固定在填料表面的逆流填料塔生物反应器。ILIUTA等[27]开发了一种逆流填充床柱反应器,将hCA Ⅱ固定在第四代高性能散堆填料上,提高了传质性能。传统的填充床柱将CA固定在填料表面,虽然提高了CO2的转化率,但其传质系数不够高,无法充分利用CA的高转化率。RASOULI等[28]提出了一种新型混合酶工艺,在填料表面和液相悬浮磁性纳米颗粒上固定hCA Ⅱ,可去除71%的CO2。
选择性膜用于去除气体混合物中的CO2。膜渗透器具有表面积大、不受重力限制等优点。但与传统化学吸收柱相比,效率较低、通过膜纤维的压差大是其主要缺点。COWAN等[29]开发了一种以CA为启动剂的含液膜(CLM)CO2捕集系统,通过在两层聚丙烯(PP)膜之间引入含磷酸盐的CA缓冲溶液,构建了CLM体系,在CA的催化下将烟气中的CO2转化为碳酸氢盐。在支撑液膜系统中,固定化液体可作为微孔膜的支撑物。FU等[30]构建了一种具有酶活性的超薄仿生膜,在环境压力和温度条件下实现CO2的捕获和分离。在含有超薄液层的纳米孔中固定CA,使CA浓度比溶液中可达到的浓度高出10倍。
在所有的CO2捕获技术中,膜接触器是最吸引人的替代技术之一,它具有接触面积大、操作灵活、液体和气体可独立控制、易于规模化和模块化等优点。RASOULI等[31]提出了一种酶促CO2捕获的新方法,在平板或中空纤维膜接触器中的膜表面和膜孔内固定CA。该生物催化膜接触器突出了固定化CA的催化效率,且在数小时内吸收率保持稳定,证实了其在工业应用中的潜力。
3 FDH催化CO2资源化
FDH在自然界中广泛存在,如厌氧和需氧细菌以及一些酵母和植物中均存在FDH。FDH可自然催化HCOO-转化为CO2(HCOO-⇌ CO2+H+2e-)。酶促反应使用各种天然辅助因子作为电子受体,特别是NAD+(辅酶Ⅰ)和NADP+。在温和条件下FDH也很容易催化反向反应,即从CO2到HCOO-,使用NADH(或NADPH)作为电子供体。这种反向F DH催化反应的实际底物是溶解的CO2还是水合的HCO3-目前尚不清楚,但最近的电化学实验已经证实,CO2是反向FDH催化的底物。FDH分为两种类型:1)活性位点含有钼(Mo)或钨(W)的金属依赖酶;2)不依赖金属驱动氧化还原催化作用的非金属依赖酶。总体而言,金属依赖型FDH相对于非金属依赖型FDH更有利于催化CO2的反向还原反应。
3.1 固定化FDH还原CO2合成甲酸
早在1984年,PARKINSON等[32]就将半导体光电极(p型磷化铟,p-InP)与生物催化剂FDH相结合,以实现CO2还原为甲酸。其后,研究人员针对此过程展开了多方面的研究,如通过固定化酶来提升酶的稳定性和重复利用率、通过改变催化条件来提高甲酸收率等。LU等[33]将FDH封装在新型海藻酸盐-二氧化硅(ALG-SiO2)杂化凝胶中,固定化FDH催化的甲酸最高产率可达95.6%,仅略低于游离形式的酶促反应(98.8%);10次循环后,固定化FDH的相对活性仍可保持在69%的较高水平。REDA等[34]报道了吸附到电极表面的含钨FDH高效催化CO2电化学还原为甲酸盐,无论是作为均相催化剂还是在电极上,FDH催化CO2还原的速率均比用于相同反应的其他已知催化剂快两个数量级以上。
表2展示了不同载体固定化FDH还原CO2合成甲酸的性能。
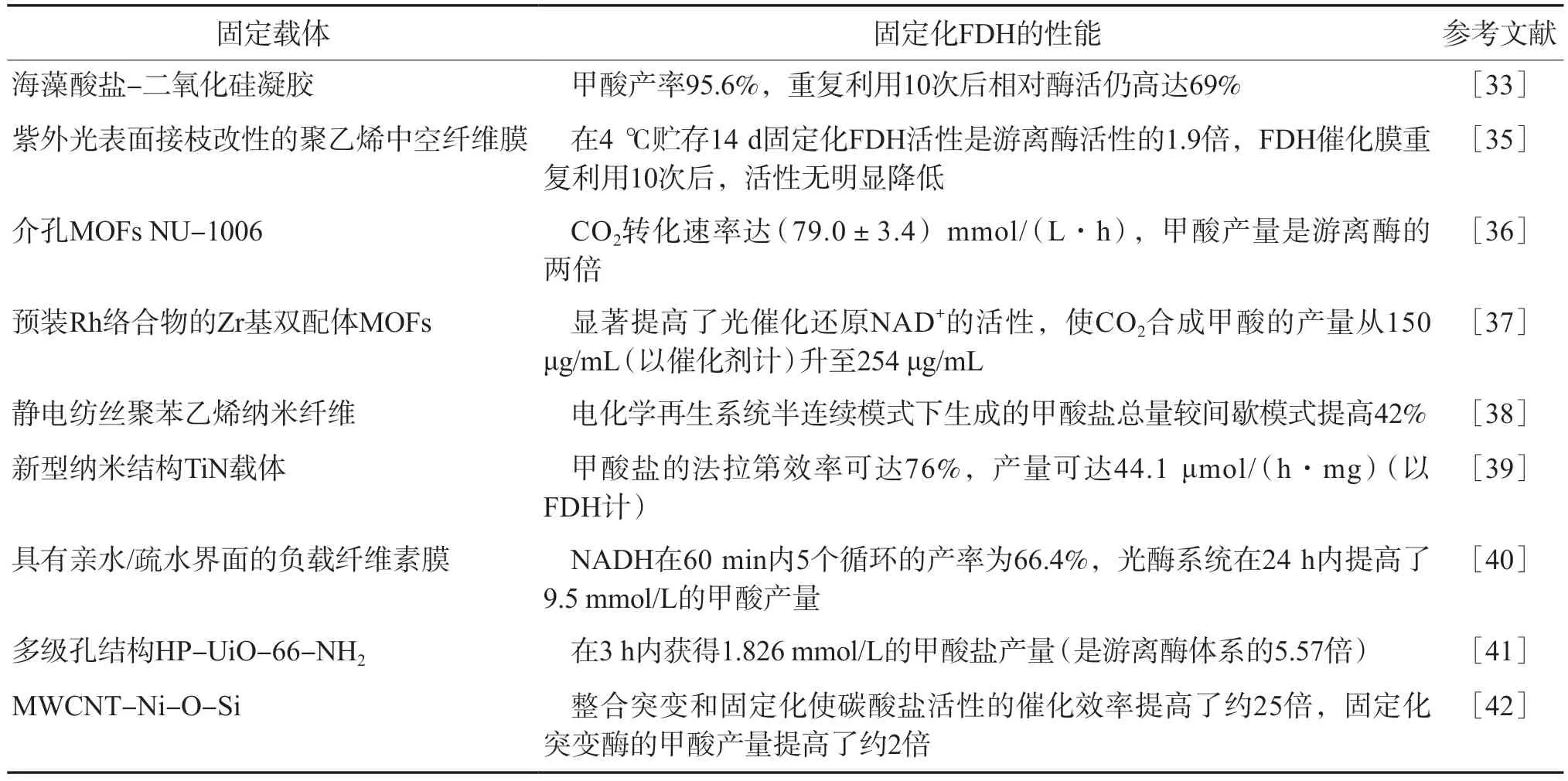
表2 不同载体固定化FDH的性能比较
MOFs材料因自身高的孔隙率和好的化学稳定性成为固定化酶的热门载体之一。XING等[37]采用具有预装Rh配合物的Zr基双配体MOFs用于NADH原位再生和FDH固定化,光敏配体四(4-羧基苯基)卟吩(TCPP)的引入增加了催化活性位点;电子介体Rh配合物锚定在Zr基双配体MOFs上,提高了电子转移效率。与UiO-66-NH2相比,Rh-H2TCPP-Ui-O-66-NH2具有优化的价带结构,显著提高了光催化还原NAD+的活性,使CO2合成甲酸的产量从150 μg/mL(以催化剂计)升至254 μg/mL。
在选择适宜FDH固定化载体的基础上,合理利用电化学技术可促进固定化FDH催化CO2转化为甲酸。ARENA等[39]构建了一个生物电化学系统(BES)用于CO2还原制甲酸,采用具有大比表面积和多级孔径分布的新型纳米结构TiN载体来固定FDH。构建的BES系统表现出显著的CO2还原性能和良好的稳定性,实现了高达76%的甲酸盐法拉第效率,最大甲酸盐产量为44.1 μmol/(h·mg)(以FDH计)。YAN等[41]将微孔UiO-66-NH2转化为同时包含微孔和介孔的多级孔结构材料(HP-UiO-66-NH2),通过优化孔结构同时增强CO2的富集和FDH的固定化,提出了酶电催化CO2还原的机理,优化后的催化体系可在3 h内获得1.826 mmol/L的甲酸盐产量(是游离酶体系的5.57倍),生成速率为6 086.7 μmol/(h·g)(以催化剂计)。
为了提升FDH的性能与稳定性,研究人员尝试通过合理设计定点或随机位点诱变不同来源的FDH。TÜLEK等[42]通过分子模拟在碳酸氢盐和甲酸盐存在下研究了Asp188突变对NAD+依赖型FDH的亚基界面附近潜在变构位点的影响,并将Asp188Arg突变型和野生型FDH固定在新合成的MWCNT(多壁碳纳米管)-Ni-O-Si载体上。整合突变和固定化使碳酸盐活性的催化效率提高了约25倍,两种固定化酶的热稳定性分别比50 ℃下的游离对应物提高了约11倍和18倍。在相同的条件下,突变酶及其固定化对应物产生的甲酸产量较野生型及其固定化对应物提高了约2倍。
3.2 以FDH为基础的多酶体系转化CO2
以FDH为基础构建的多酶体系转化CO2越来越受到关注。ILIUTA等[43]提出了在固定床微反应器(FBMR)中,利用共固定化耐氧FDH和葡萄糖脱氢酶,通过酶介导的CO2还原过程将大气中的CO2转化为甲酸盐,同时原位再生NADH。由于该系统增强了相间传质和气液界面面积,CO2转化率明显提高。因此,具有固定化FDH和葡萄糖脱氢酶的FBMR在CO2绿色转化方面有着独特优势。
FDH、甲醛脱氢酶(FADH)和甲醇脱氢酶(MDH)构成的多酶体系转化CO2的研究较多。1999年,OBERT等[44]通过由3种脱氢酶催化的级联反应将CO2还原为甲醇,利用NADH充当每个脱氢酶催化还原的末端电子供体,获得了91.2%的甲醇收率。自此,甲醇一直是CO2催化转化的重点研究方向。XU等[45]将FDH、FADH和乙醇脱氢酶(ADH)封装于ALG-SiO2杂化凝胶中,甲醇收率可达98.1%,储存60 d后收率仍可达76.2%,在10次循环后还可达78.5%。姜忠义等[46]以正硅酸乙酯为前驱体,用改进的溶胶-凝胶法对3种酶进行了包埋共固定化,在低温低压下将CO2转化为甲醇,甲醇收率可达92.4%。SUN等[47]通过简单温和的仿生矿化过程,将3种脱氢酶包裹在二氧化钛颗粒中,建立了一个绿色高效的多酶系统,该系统可有效地将CO2转化为甲醇,在更宽的pH和温度范围内获得了更高的反应收率。EL-ZAHAB等[48]把FDH、FADH和ADH共固定在聚苯乙烯微粒上,将CO2转化为甲醇,固定多酶体系11次循环使用后产生了48%的累计甲醇收率。
COCUZZA等[49]研究了OBERT等提出的级联反应的第一步,使用与甘油脱氢酶(GlyDH)共固定的FDH将CO2生成甲酸。通过GlyDH将甘油氧化为二羟基丙酮,以还原形式再生烟酰胺辅因子。在以斜发沸石作为载体时,FDH的保留活性大幅增加,从18%升至89%。FDH和GlyDH的固定化产率约为100%。研究表明,该方法可能在酶固定化和生物催化方面得到进一步发展。
4 CA与FDH的级联反应体系
从能量角度而言,甲酸是利用CO2的最经济的方法。以FDH为生物催化剂,NADH为辅因子,CO2可逆地转化为甲酸。然而,该反应在热力学上是不利的,逆向反应的速率常数远高于正向反应,并且在环境压力下,CO2在水相中的溶解度较低。为了促进反应,提高反应溶液的CO2吸收是一种有效策略。CA能显著加速CO2的水合,在CO2分离捕集方面具有重要的应用前景。
JI等[50]通过将FDH、FADH、ADH和谷氨酸脱氢酶(GDH)4种酶和辅因子原位包埋在阳离子聚电解质掺杂的中空纳米纤维腔内,并通过在中空纳米纤维的外表面上组装CA加速CO2水合,CO2还原酶系统实现了103.2%的最高甲醇收率,在10次循环后,仍保留了约80%的初始活性,基于辅因子的累积甲醇产率达940.5%。WANG等[51]研究了CA对催化FDH中甲酸盐合成的影响,优化各种参数后反应速率提高了4.2倍。SATO等[52]利用FDH和CA构建了双酶系统,证明碳捕集、利用与封存(CCUS)系统中使用CA将CO2转化为碳酸氢盐和使用FDH将CO2还原为甲酸盐是协调的。ZHANG等[53]通过微生物谷氨酰胺转胺酶(MTG)的“交联介质”作用将CA和FDH连接在一起,以获得不同比例的“一对一”和“一对多”的交联酶聚集体,交联后CA的保留酶活性超过93%,FDH的保留酶活性超过84%;与游离酶相比,交联酶的总催化效率提高了5.8倍,并且FDH在不同温度下的热稳定性得到改善。
在级联反应体系中引入固体吸附剂是加强CO2转化的策略之一,其中具有高胺基密度的聚乙烯亚胺(PEI)作为聚合物改性剂起着至关重要的作用。将CA和FDH共固定在PEI上优势明显,CA和PEI均可极大地促进CO2的捕获和HCO3-的形成,从而加速FDH催化转化CO2为甲酸盐。同时,酶在固体吸附剂上的表面附着使其易于分离和回收,并具有较高的稳定性。ZHAI等[54]把CA固定在PEI和聚多巴胺(PDA)改性的SiO2微球(PDA/PEI-SiO2-CA)上,并将其加入到含有NADH和FDH的反应溶液中,促进CO2转化为甲酸盐,在PDA/PEI-SiO2-CA存在下,初始反应速率是空白对照的48.6倍,甲酸盐产量也高于空白对照。介孔二氧化硅(mSiO2)材料因其大比表面积、高浓度表面硅羟基以及出色的化学/热稳定性受到研究人员的关注。MAO等[55]制备了结构可控的mSiO2纳米颗粒并用PDA和PEI对其进行改性,用于CO2捕获和转化为甲酸的集成过程。mSiO2本身就是一种强CO2吸附剂,与游离酶相比,添加0.01 g mSiO2(410)(粒径410 nm)可使CO2转化加速11.94倍。用0.05g PDA/PEI-mSiO2(340)和PDA/PEI-mSiO2(410)改性后,酶催化反应较无颗粒系统加速了24.0倍和30.8倍,以PDA/PEI-mSiO2(410)为载体进行FDH和CA的共固定化,在4 ℃下使用10次后仍可保持86.7%的活性,21 d后可保持55.2%的活性(游离酶仅保持29.6%的活性)。
沸石咪唑酯骨架材料ZIF-8的咪唑基团能够协同催化CO2的水合反应。REN等[56]通过将CA、FDH、辅因子NADH和GDH同时封装到ZIF-8中,构建了纳米级多酶反应器。在多酶反应器中,FDH的活性回收率达50%,CA和ZIF-8的组合显著促进了CO2在反应溶液中的溶解。纳米级多酶反应器表现出优异的CO2转化为甲酸盐的能力,与游离多酶体系相比甲酸盐的产率提高了4.6倍,在8次循环后仍保持了50%的初始活性,具有良好的可重复使用性。WANG等[57]以ZIF-8为模板,制备了具有分级结构和孔隙率的超薄PDA微胶囊,将CA、FDH和GDH封装在PDA微胶囊中。微胶囊中FDH保留了94.7%的等效游离FDH的活性。与游离多酶相比,包埋在PDA微胶囊中的固定化多酶表现出4.5倍的甲酸盐产量。CHAI等[58]合成了包埋两种酶(CA和FDH)的ZIF-8薄膜,该ZIF-8生物催化膜在沸水中处理1 h后仍表现出优异的热稳定性;在气液膜接触器中催化4 h后甲酸产率为22%;催化活性较无ZIF-8时增加了1.6倍。
5 结语与展望
生物酶技术固定和转化CO2既可缓解温室效应带来的环境问题,又能以温室气体为碳源合成高附加值产品,变废为宝,有望发展成一种可循环的CO2管理体系。然而,现阶段生物酶技术固定并转化CO2仍存在一些问题:一方面,现有的生物固碳途径反应过程大都较长,固碳酶尚存在缺陷;另一方面,在酶法催化CO2捕集转化过程中,关键酶的核心驱动力不足,限制了反应速率,而其较差的稳定性是应用于工业生产的又一障碍。为了CO2捕集和利用的进一步发展,未来的生物酶技术可从以下方面进行研究:
a)研发更高效的核心固碳酶,强化CO2羧化途径,优化能量供给,构建全新的人工固碳途径;
b)探索开发廉价高效的固定支撑材料,研发创新和有效的固定化技术,以保持酶的活性和稳定性;
c)剖析酶级联反应体系的热力学和动力学限制因素,从级联反应的原理着手,提高反应效率。
猜你喜欢
中成药(2021年5期)2021-07-21
中学生数理化·七年级数学人教版(2019年10期)2019-11-25
上海包装(2019年2期)2019-05-20
中学生数理化·高一版(2018年9期)2018-10-09
湖南教育·C版(2018年3期)2018-06-05
中成药(2018年2期)2018-05-09
中国蜂业(2018年4期)2018-05-09
当代化工研究(2016年6期)2016-03-20
微生物与感染(2015年1期)2015-02-28
无机化学学报(2014年3期)2014-02-28
