
一步净化结合超高液相色谱串联质谱同时测定雅鱼中13 类54 种兽药残留
2024-01-30 02:12:12杨巧慧刘中良李霞雪张建雄闻瑞琪
食品工业科技 2024年3期
杨巧慧,刘中良,陈 亚,李霞雪,张 宇,曾 艳, ,张建雄,闻瑞琪,兰 韬
(1.雅安市农产品质量监测检验中心,四川雅安 625000;2.唐山市食品药品综合检验检测中心,河北唐山 063000;3.北京科德诺思技术有限公司,北京 102299;4.中国标准化研究院,北京 100191)
“雅鱼”(Schizothorax prenanti)又名丙穴鱼,四川裂腹鱼。属于硬骨鱼纲、鲤形目、鲤科,裂腹鱼亚科,包括齐口裂腹鱼、重口裂腹鱼、等一大类裂腹鱼[1],主要分布在长江支干流中。近年来,由于大修水利工程、水体环境污染及过度捕捞等原因,野生“雅鱼”数量急剧减少,“雅鱼”已列入长江上游急需保护的特种鱼类[2]。目前,人工养殖“雅鱼”已在四川、云南、贵州等地广泛分布,经济效益显著[3-5]。特别是四川雅安养殖的齐口和重口裂腹“雅鱼”已被评为国家地理标志产品[6-7]。随着人工养殖规模的扩大,在养殖过程中,常会出现败血症、烂鳃病、水霉病等鱼病,因此使用磺胺类或其它抗生素、抗菌剂等进行预防和治疗。但部分养殖者为提高经济效益或缺乏对药物危害的认知而不合理用药,甚至非法滥用违禁药物。这些药物会通过食物链给人们带来危害,因此,养殖雅鱼的质量安全日益受到关注。目前,水产品中的兽药残留检测方法主要有薄层色谱法(TLC)[8]、液相色谱法(HPLC)[9]、液相色谱串联质谱法(LC-MS/MS)[10-11]等。其中,LC-MS/MS 具有高分辨率、高通量的优点,已被广泛应用于兽药残留检测中[12-13]。
水产品中兽药残留的检测最具挑战性的步骤是提取、净化样品[14]。在现有的前处理手段中,主要的净化方式有固相萃取法(Solid phase extraction,SPE)、液液萃取法(Liquid-liquid extraction,LLE)以及近年来被广泛应用的快速、简单、廉价、高效、安全的前处理方法(Quick easy cheap effective rugged and safe methodology,QuEChERS)[15]等。与前两种净化方式相比较,QuEChERS 法具有使用溶剂少、高样品通量、操作简单、成本低廉等优势,已成为对兽药残留分析具有很强适用性的前处理方法[16-17],该方法较传统的SPE 前处理式更为简单,但仍需多次离心、多次转移液体。但同时也有研究发现,传统QuEChERS 方法中提取剂内含有少量水,加入无机盐后,通过离心分层为有机相和水相,然后再分散固相萃取进行样品净化,虽然这种方法提高了非极性兽药的提取率,但盐的分配却会导致金属与四环素等化合物的螯合作用,导致分析物损失[18-19]。Han 等[20],Lehoyay 等[21]研究发现,QuEChERS 方法中水相的加入,导致部分强极性目标物保留在水相中,提取时不能完全转移到有机相中,从而获得较差的提取效率。
方静等[22]研究表明四川野生“雅鱼”中含有较高的蛋白质(含量高达16.72%),赖氨酸含量甚至高出鸡蛋蛋白质评分模式的50%。但罗润博等[23]、张洁等[24]研究发现,蛋白质不仅会干扰待测物的测定,还会引起仪器进样口堵塞等问题,因此前处理过程中需将蛋白质进行沉淀。赵燕英等[25]的研究表明裂腹鱼(雅鱼)背部与腹部肌肉的肌内脂肪(Intramuscular fat,IMF)含量极显著高于其他鱼类,这也增加了样品的净化难度。
本试验在QuEChERS 法基础上采用增强除脂、蛋白一步净化的方式(Clean-up LPAS 法,lipid and protein adsorbent)这是一种改良的QuEChERS 方法,通过在吸附材料表面修饰,有针对性地吸附脂肪及蛋白,最大限度地减轻目标药物的基质效应。同时方法还打破了传统分散萃取、固相萃取的方式,提取时无需加入无机盐,净化只需一步即可,结合LCMS/MS 的检测对“雅鱼”中22 种磺胺类、10 种喹诺酮类、4 种酰胺醇类、4 种四环素类、3 种大环内酯类、2 种镇静类、2 种林可胺类、1 种β-内酰胺类、1 种皮质类激素、1 种抗球虫类、1 种抗病毒类、1 种硝基咪唑类以及孔雀石绿及隐色孔雀石绿共计13 类,54 种兽药残留检测分析进行研究,以期建立快速、准确、高效、经济检测“雅鱼”中多种兽用药物残留的方法,为“雅鱼”这一地理标志产品的健康养殖、用药监管和民众舌尖上的安全提供保障。
1 材料与方法
1.1 材料与仪器
甲醇、乙腈、甲酸 色谱纯,赛默飞世尔科技(中国)有限公司;乙酸铵(色谱纯)、乙二胺四乙酸二钠(EDTA 二钠)(分析纯)天津市科密欧化学试剂有限公司;KNORTH Clean-up LPAS 净化柱 北京科德诺思技术有限公司;实验用一级水 均为本实验室Milli-Q 型超纯水仪所制;所有标准物质(含内标物质)均为纯度大于95%的固体标准物质,购自德国 Dr.Ehrenstorfer 公司,标准物质名称、CAS 号如表1 所示;“雅鱼”样品 来自雅安市“雅鱼”养殖基地,随机采样。
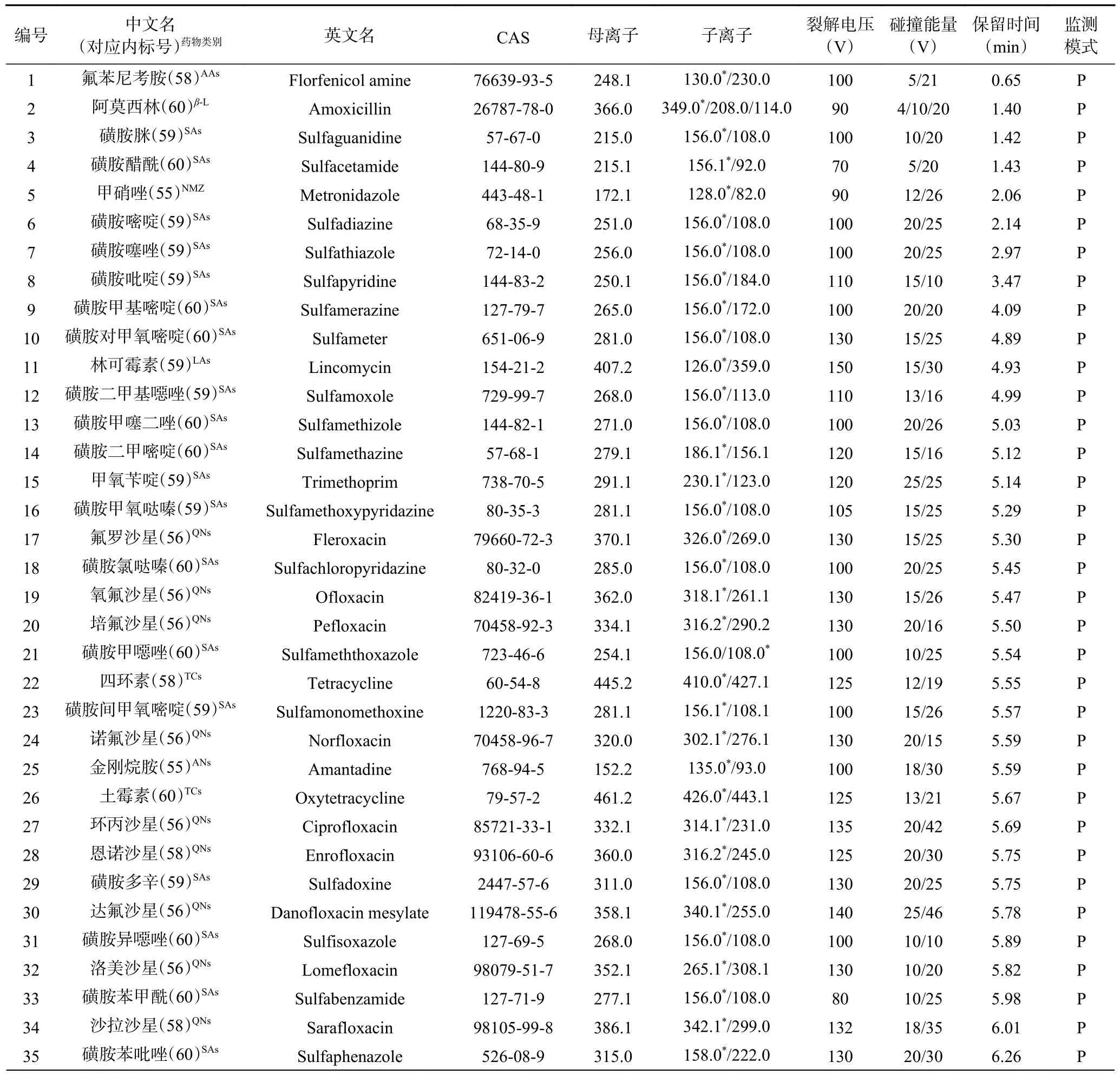
表1 目标化合物名称及质谱采集条件Table 1 Name of each target compound and the MS acquisition conditions
1290-6470 型液相色谱串联质谱仪,配有电喷雾离子源(ESI)美国安捷伦公司;TGL-16 型高速冷冻离心机 四川蜀科仪器有限公司;KN-026S 型强力多管漩涡混合仪 北京科德诺思技术有限公司;PD500 型高速分散匀浆机 英国 Prima Technology Group;GD16Plus 型高速研磨仪 深圳新锐科技有限公司;KH-500B 型超声波清洗器 昆山禾创超声仪器有限公司;Milli-Q 超纯水机 美国 Millipore 公司。
1.2 实验方法
1.2.1 样品制备与保存 “雅鱼”样品的抽取参考GB/T 30891-2014[26]水产品抽样规范进行,制备和保存参考NY/T 3304-2018[27]中水产品类样品制备与保存方法进行。将鲜活“雅鱼”宰杀后去除头、骨、内脏等组织,取其肌肉、鱼皮组织切块,通过组织捣碎机进行粉碎,混匀,分装入密实袋中,贴好标签,于-20 ℃条件下冷冻保存。
1.2.2 标准溶液配制 准确称取各目标组分10.00 mg标准物质于10 mL 棕色容量瓶中,用甲醇充分溶解后定容至刻度,混匀后转移至棕色标样瓶中,配制成浓度为1 mg/mL 的单一标准储备液,-20 ℃保存。其中,阿莫西林用乙腈:水(1:1)溶解和定容;喹诺酮类药物先用适量0.03 mol/L 的氢氧化钠溶液溶解,然后用甲醇稀释定容至刻度。随后将所有单标用甲醇稀释配制成浓度为10 μg/mL 的混合标准溶液液,将该混合标准溶液用超纯水或基质稀释配制成浓度为0.1、0.2、0.5、1.0、2.0、5.0、10.0、20.0、50.0 μg/L的系列标准工作曲线。
1.2.3 样品前处理 称取(2.00 ±0.05)g 样品于50 mL具塞离心管中,准确加入0.2 μg/mL 混合内标溶液100 μL,涡旋混匀,避光静置20 min。准确加入8 mL 0.2%甲酸的乙腈:水溶液(90:10),2000 r/min涡旋振摇2 min 后冰水浴超声20 min 提取,于5000 r/min,4 ℃下离心5 min。取上清液2 mL 过LPAS 净化柱,净化柱无需活化、洗脱等步骤,只需在重力作用下通过净化柱,一步收集滤液。准确移取0.4 mL 滤液,加入0.6 mL 稀释液(5 mmol/L 的乙酸铵溶液)于同一离心管中,涡旋混匀,过0.22 μm×13 mm PTFE 有机滤膜至2 mL 进样瓶,供LC-MS/MS 仪器分析。
1.2.4 LC-MS/MS 条件 色谱条件:色谱柱:Agilent Zorbax SB-C18,50 mm×0.30 mm,1.8 µm;进样量2.0 µL;流动相A 为甲醇,B 为水相(0.1%甲酸/5 mmol/L 乙酸铵溶液);梯度洗脱程序:0~2 min,10%A,90%B;2~7 min,10%~80%A,90%~20%B;7~7.5 min,80%A,20%B;7.5~8 min,80%~95%A,20%~5%B;8~9 min,95%A,5%B;9~9.5 min,95%~10%A,5%~90%B;9.5~14 min,10%A,90%B;柱温35 ℃;流速:0.3 mL/min;
质谱条件:电喷雾电离(ESI)源,正、负离子模式;干燥气温度,325 ℃;毛细管干燥气流量(Gas Flow):7 L/min;雾化器压力(Nebulizer):35 psi;鞘气流量(Sheath Gas Flow):12 L/min;鞘气温度(Sheath Gas Heater):300 ℃;扫描方式为动态多反应监测(dMRM),内标法标准曲线定量。各目标化合物的质谱采集条件如表1 所示。
1.3 数据处理
采用Agilent LC-MS QQQ MassHunter 采集软件进行数据采集,LC-MS QQQ MassHunter 定量软件进行数据分析,包括标准曲线的建立、结果计算,处理数据用Excel 2019 处理后用Origin Pro 9.0 绘制图形。本试验前处理条件优化均采用三个平行样品进行,外标法进行计算。前处理条件优化完成后,方法学验证采用内标法进行计算。
2 结果与分析
2.1 仪器条件优化
为保证每个参数都能顺利出峰,且得到较为理想的峰型,试验反复调整了流动相的组成及梯度洗脱程序等关键性液相参数。重点比较了甲醇和乙腈作为有机相与0.1%甲酸水、0.1%甲酸/5 mmol/L 乙酸铵混合溶液的不同组合、比例,同时,不断调整流动相梯度,确保每个参数在无干扰的情况下在较短的时间内正常出峰。通过试验,综合各目标化合物的峰型、检测灵敏度以及同分异构体分离度,最终确定本试验最佳流动相为甲醇-0.1%甲酸/5 mmol/L 乙酸铵溶液,梯度洗脱条件如1.2.4 中所示。
在优化好的色谱条件下,用单一标准溶液(2 mg/L)对母离子进行全扫描(MS2Scan),同时设置几组不同的裂解电压(Frangment/V),对照每种目标物的相对分子量,找到母离子,选择母离子响应最大值下的裂解电压,作为后续最优裂解电压。产物离子扫描(Product ion scan)时优化碰撞电压(CE/V),找到特征子离子及最佳CE,保证除内标物外的每个参数均至少有两个特征离子对。最后采用优化好的条件,进行动态多反应监测(dMRM)分析,内标法标准曲线定量。54 种兽药组分的保留时间、MRM 离子对、监测模式及最佳CE 见表1,MRM 总离子图见图1、图2。
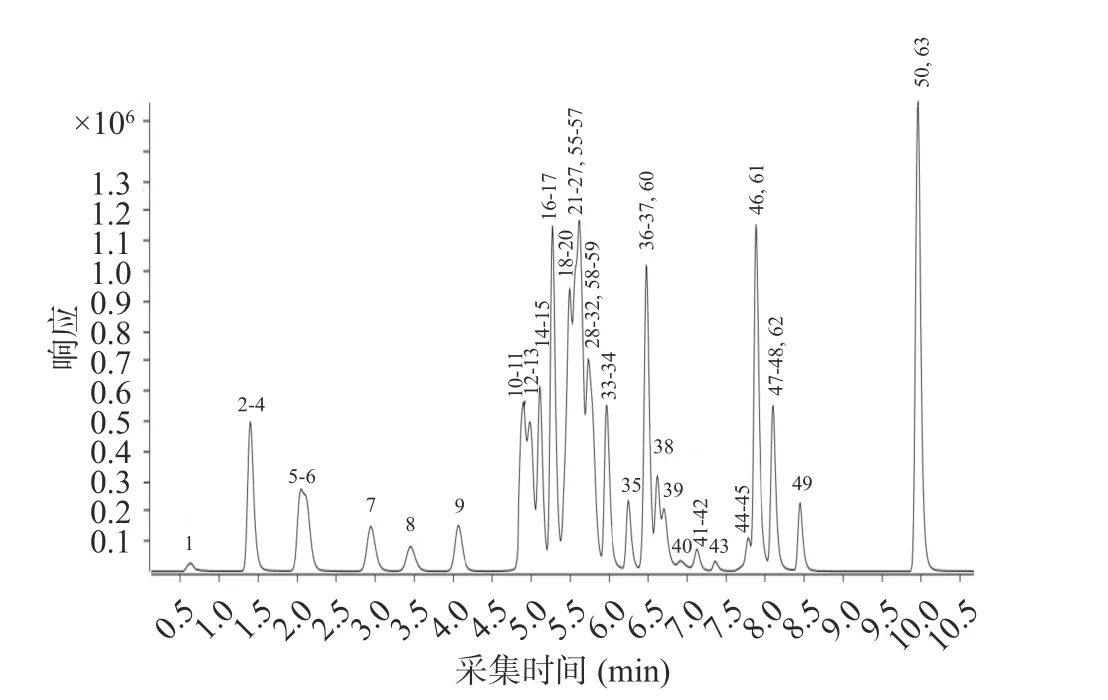
图1 正离子模式下MRM 总离子图(10 μg/L)Fig.1 Total MRM ion diagram in positive ion mode (10 μg/L)

图2 负离子模式下MRM 总离子图(10 μg/L)Fig.2 Total MRM ion diagram in negative ion mode (10 μg/L)
2.2 前处理条件优化
2.2.1 提取方式 兽药残留检测中常用的提取方式有超声、研磨、均质、涡旋等。试验比较了冰水浴超声20 min、研磨(2000 r/min,120 s)、均质(8500 r/min,1 min)三种提取方式对目标物提取效果的影响,不同提取方式的回收率结果如图3。由图可知,这三种提取方式的提取效果基本一致,但超声的提取效果相对较好,54 种目标物有46 种目标物的回收率在60%~120%之间,优于研磨提取的43 种、均质提取的41种,因此选择超声提取进行后续试验。

图3 不同提取方式对提取效果的影响Fig.3 Effect of different extraction methods on the extraction effect
在此基础上,试验还发现常温超声及冰水浴超声对目标物提取效果的影响(试验在7 月进行,常温超声后水温约为40~50 ℃),回收率结果如图4 所示,结果表明,常温超声后,有55.56%的目标物未检出,有11.11%的目标物回收率低于60%;采用冰水浴超声时,有85.19%的目标物的回收率在60%~120%之间。结果表明超声温度对目标物的提取影响较大,因此,在进行超声提取时采取冰水浴进行超声。
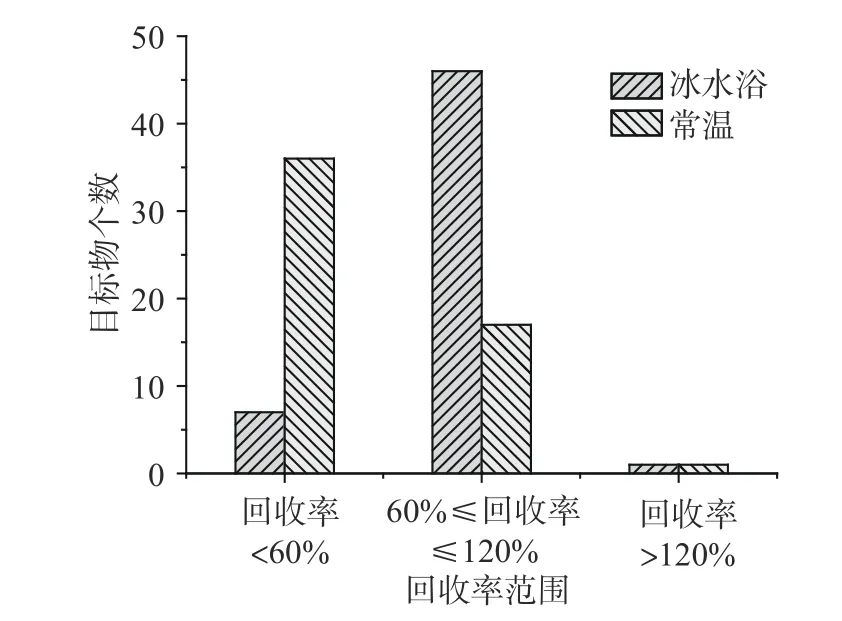
图4 超声温度对提取效果的影响Fig.4 Effect of ultrasound temperatures on the extraction effect
2.2.2 提取体积 通过对目标物进行添加回收试验,比较4、6、8、10 mL 提取剂对提取效果的影响,回收结果如图5A、5B 所示。结果表明,在提取液为4、6、8 mL 时,大部分目标物的回收率随提取体积的增加而趋于100%。当提取体积为10 mL 时,目标物的回收率大于120%的个数开始增加,因此试验选择提取体积为8 mL。

图5 不同提取体积回收率情况Fig.5 Recovery of different extraction volumes
2.2.3 缓冲盐选择 本试验比较在提取液中加入EDTA 二钠(1 g)与不加入对目标物的提取影响,结果发现仅对四环素类(四环素、土霉素、金霉素、强力霉素)药物影响较大(图6),与Zhao 等[28]的研究结果一致,这可能是土霉素、金霉素、强力霉素易与金属离子形成螯合物的原因[29]。整体回收率在60%~120%之间,为节约成本,本试验选择不加入EDTA 二钠,也不加入其它盐类进行提取。

图6 EDTA 二钠对提取效果的影响Fig.6 Influence of EDTA-2Na on extraction effect
2.3 基质效应
基质效应(Matrix effects,ME)是由于基质中其他干扰物质的存在,导致被分析物的信号强度有不同程度的增强或减弱的现象[30]。当采用LC-MS/MS分析复杂的样本时,在大气压电离(Atmospheric pressure ionization,API)界面下,干扰物质与目标分析物共洗脱时,它们可以改变源中的电离效率,可能导致电离抑制或电离增强,这会直接影响定量的准确性,除非基质效应被最小化或补偿[31-32]。
本试验用空白基质溶液配制的标准曲线斜率(k1)与试剂标准曲线斜率(k2)进行比较(ME=k1/k2),ME>1 为基质增强,ME<1 为基质减弱,ME=1 为没有基质效应,ME 在0.8~1.2 之间,为可接受范围[33]。试验比较了54 种兽药在“雅鱼”中的基质效应,结果如表2 所示。
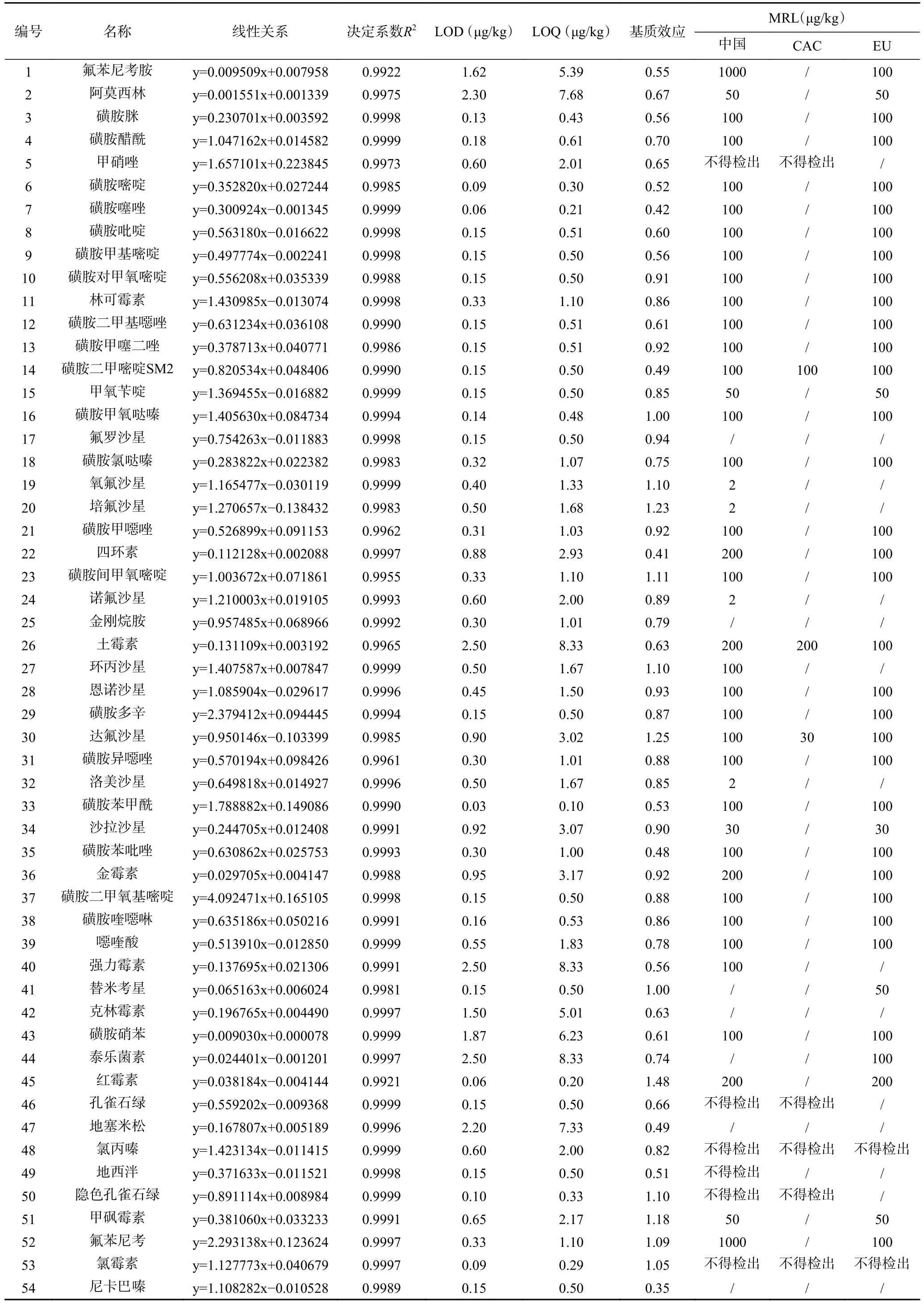
表2 54 种兽药的方法验证结果及基质效应、限量值Table 2 Method validation results and matrix effects and limit values of the 54 veterinary drugs
由表2 可知,有26 种兽药ME<0.8,3 种兽药ME>1.2,25 种兽药ME 在0.8~1.2 之间,表现为可接受。由此可见,“雅鱼”中的脂肪、蛋白质、氨基酸等物质对试验中53.7%的兽药存在基质干扰。因此,为对基质效应进行有效补偿,本文采用样本空白基质结合内标物的方式进行校正,通过加标回收试验验证,结果可靠。
2.4 方法验证
2.4.1 方法检出限、方法定量限及线性范围 各兽药及其代谢物的验证参数见表2。以目标化合物的响应值为纵坐标,质量浓度为横坐标,绘制标准工作曲线。结果显示,54 种兽药在0.1~50 μg/L 范围内线性良好,决定系数(R2)均在0.992 以上。在空白基质中添加不同水平的目标化合物(5、10、20、50 μg/kg)。方法检出限(LOD)和方法定量限(LOQ)根据不同添加水平的标准偏差(SD)与斜率(k)的比值计算,LOD=3.3×(SD/k),LOQ=10×(SD/k)[34]。
54 种兽药在“雅鱼”中的LOD 为0.03~2.5 μg/kg,LOQ 为0.1~8.3 μg/kg,基本优于现有国标中LOD、LOQ[35-37]。各兽药在中国[38-39]、食品法典委员会(CAC)[40]、欧盟(EU)[41]的限量值(MRL)见表2,其中有MRL 值的兽药在本试验中的LOD 值均低于其MRL 值,表明该方法的方法检出限较低、灵敏度较高,能满足现有检测的需求。
2.4.2 准确度及精密度验证 通过对空白样品中加入低、中、高三个水平,其中,对LOQ≤5 μg/kg 的目标物添加量为5、10、50 μg/kg,对LOQ>5 μg/kg 的目标物添加量为10、20、50 μg/kg,每个水平三平行样做加标回收试验,计算平均回收率及相对标准偏差(RSD),验证本方法的准确度和精密度。结果表明(表3),在低、中、高三个加标水平下54 种兽药的回收率在70.45%~118.10%之间,RSD 在0.01%~9.85%之间,符合方法验证对准确度和精密度的要求[33,42],能满足“雅鱼”中54 种兽药残留要求。
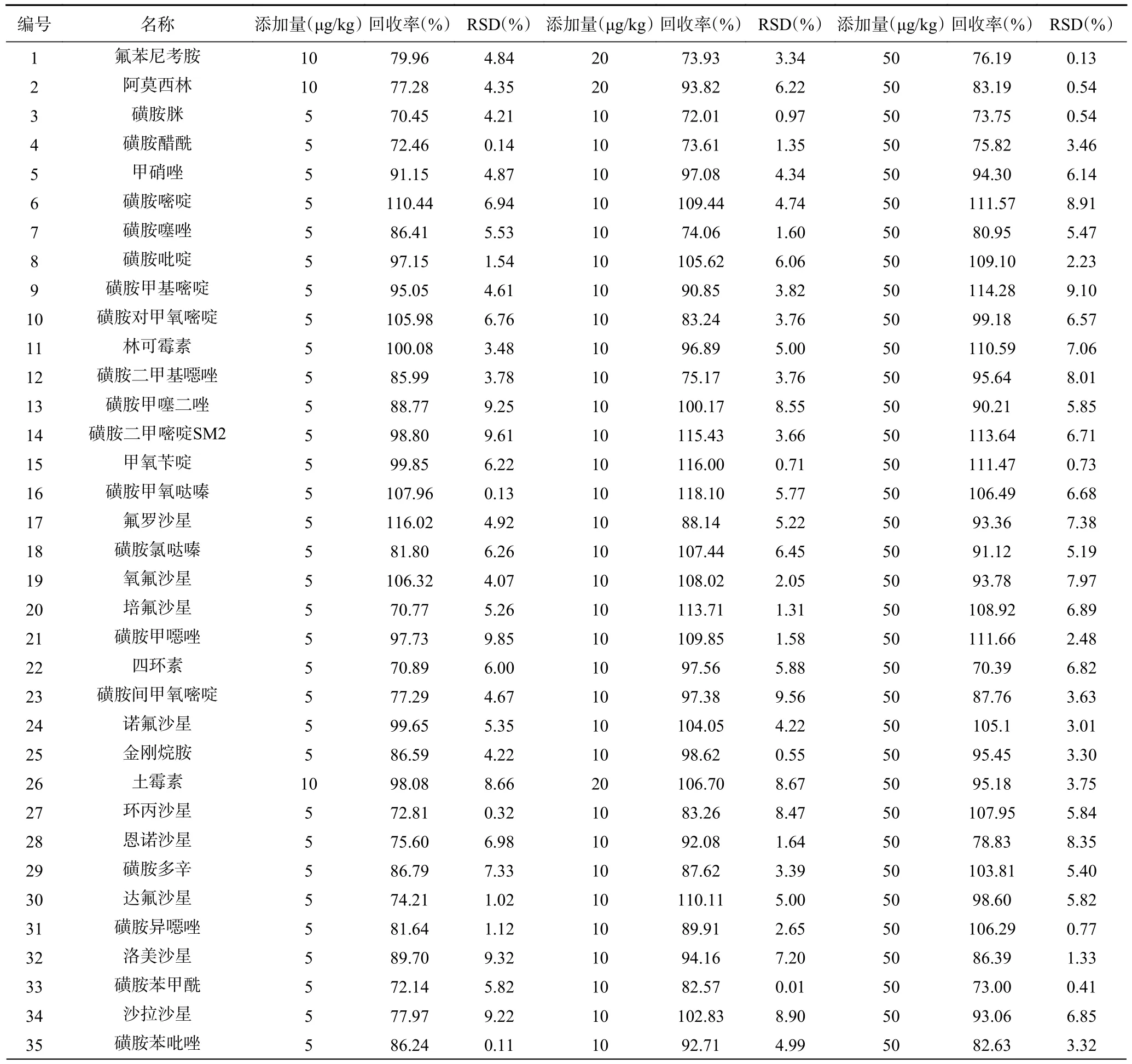
表3 54 种兽药不同添加量的回收率及精密度Table 3 Recovery rate and precision of different added amounts of 54 veterinary drugs
2.5 实际样品检测
应用试验建立的方法对2022 年实验室随机抽取养殖基地50 份雅鱼样品进行检测。结果显示,有7 份样品有药物检出,其中,恩诺沙星检出2 次(检出值分别为0.98、0.58 μg/kg),达氟沙星各检出2 次(检出值分别为1.52、1.32 μg/kg),磺胺甲噁唑检出4 次(检出值分别为0.54、0.63、0.47、0.88 μg/kg),共计8 次。以上检出药物均低于该方法定量限,远远低于表2 中限量值(均为100 μg/kg),其它药物均无检出,所有样品均为合格样品。
3 结论
试验对检测仪器条件和前处方法检出限理条件(提取方式、提取剂体积、缓冲盐)进行了优化比较,在传统QuEChERS 法的基础上建立了利用一步净化结合液相色谱串联质谱仪,同时对养殖“雅鱼”中13 类54 种兽药残留进行检测的高通量检测方法。本方法与传统QuEChERS 法相比,提取剂内不含水,因此,减少了盐析、液体转移、离心等步骤,净化后液体较QuEChERS 净化液清澈,操作更为简单、快捷。利用空白基质结合内标定量的方式,使结果更稳定、可靠。利用“雅鱼”基质进行了低、中、高三水平的加标回收,对准确度和精密度、线性范围、及基质效应等进行了方法验证,结果均符合实验室质量控制规范的要求。利用该方法对50 份养殖“雅鱼”样本进行检测,共7 份样品,8 样次定性检出。由此可见,“雅鱼”在养殖过程中确实有使用喹诺酮类和磺胺类药物对其进行鱼病治疗,但均未超出限量值,总体质量安全,但后期应加强监测,加强兽药使用指导和宣传。
综上所述,本研究建立了一种可以快速、准确、高通量且经济检测“雅鱼”中多种类药物残留的快速定性定量方法,为后续加大对“雅鱼”养殖用药的监管和药物残留的检测,持续保证“雅鱼”的质量安全提供了有效的技术支持。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23 06:22:32
中国土壤与肥料(2021年5期)2021-12-02 01:04:34
今日农业(2020年22期)2020-12-14 16:45:58
当代水产(2019年3期)2019-05-14 05:42:48
Coco薇(2017年7期)2017-07-21 16:49:50
河南畜牧兽医(2016年24期)2016-11-29 01:28:18
金色年华(2016年23期)2016-06-15 20:28:28
兽医导刊(2015年7期)2016-01-04 11:59:54
电力需求侧管理(2014年4期)2014-03-20 13:35:51
云南畜牧兽医(2014年4期)2014-02-28 21:25:33
