
手性磷杂环化合物合成研究进展
2024-01-23 12:54涂星宇苗志伟
大学化学 2023年11期
涂星宇,苗志伟
南开大学化学学院,元素有机化学国家重点实验室,天津 300071
有机磷化学在化学领域中占有重要地位,已经发展成为一门独立的学科方向。含磷有机化合物在农药、医药、催化、萃取、阻燃剂和燃料固化阻滞剂等诸多领域均有广泛应用[1]。手性磷杂环化合物是一类重要的有机磷化合物,其作为金属配体或手性小分子催化剂在不对称合成研究领域占有重要地位,受到了有机化学家的广泛关注[2]。图1中所示的手性磷杂环化合物A和B可以分别作为金属钯和金属铱的配体,对烯烃的不对称氢化反应起到催化作用[3,4];化合物C则可以作为金属镍的配体实现炔和醛的不对称交叉偶联反应[5];化合物D是一种手性磷杂环催化剂,可以催化二芳基氧膦衍生物的不对称动力学拆分反应[6]。正因为手性磷杂环化合物在不对称合成领域中的重要价值,使得该类化合物的高效构建方法一直是有机磷化学中非常活跃的研究领域,本文综述了近年来手性磷杂环化合物合成研究进展。
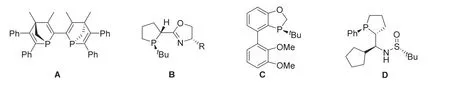
图1 作为金属配体或小分子催化剂的手性磷杂环化合物
1 利用过渡金属催化不对称合成手性磷杂环化合物
过渡金属催化是合成手性化合物的一种重要方法,其具有手性原料经济性、合成方式多样性以及反应高效性等优点,是一种应用非常广泛的不对称合成手段[7]。因此,在磷杂环化合物不对称合成的诸多方法中,过渡金属催化法是最受关注的方法之一,目前已经报道了钼、钯、铑、金、钴等多种过渡金属用来催化合成手性磷杂环化合物的反应。
2008年,Hoveyda课题组[8]将烯烃的不对称闭环复分解反应(ARCM)拓展到了手性磷杂环的不对称合成上。作者以非手性的多烯烃次膦酸酯1为底物,在钼催化剂2的作用下在60 °C中反应13 h,能以79%的收率和96%的ee值获得手性七元磷杂环化合物3 (图2)。通过调整次膦酸酯结构,也能同样以高收率和ee值获得五元和六元手性磷杂环。该反应机理与常规烯烃复分解反应的机理类似,钼催化剂与底物发生配体交换后生成的金属卡宾络合物4与底物上的双键发生[2+2]环加成反应得到中间体5,随后经过逆环加成反应生成目标产物3。基于这一机理,通过控制钼催化剂配体的空间位阻,可以定向地得到特定磷手性的目标产物。该方法成功在含磷的多烯烃结构上应用了ARCM反应,为手性磷杂环化合物的合成提供了新的思路。
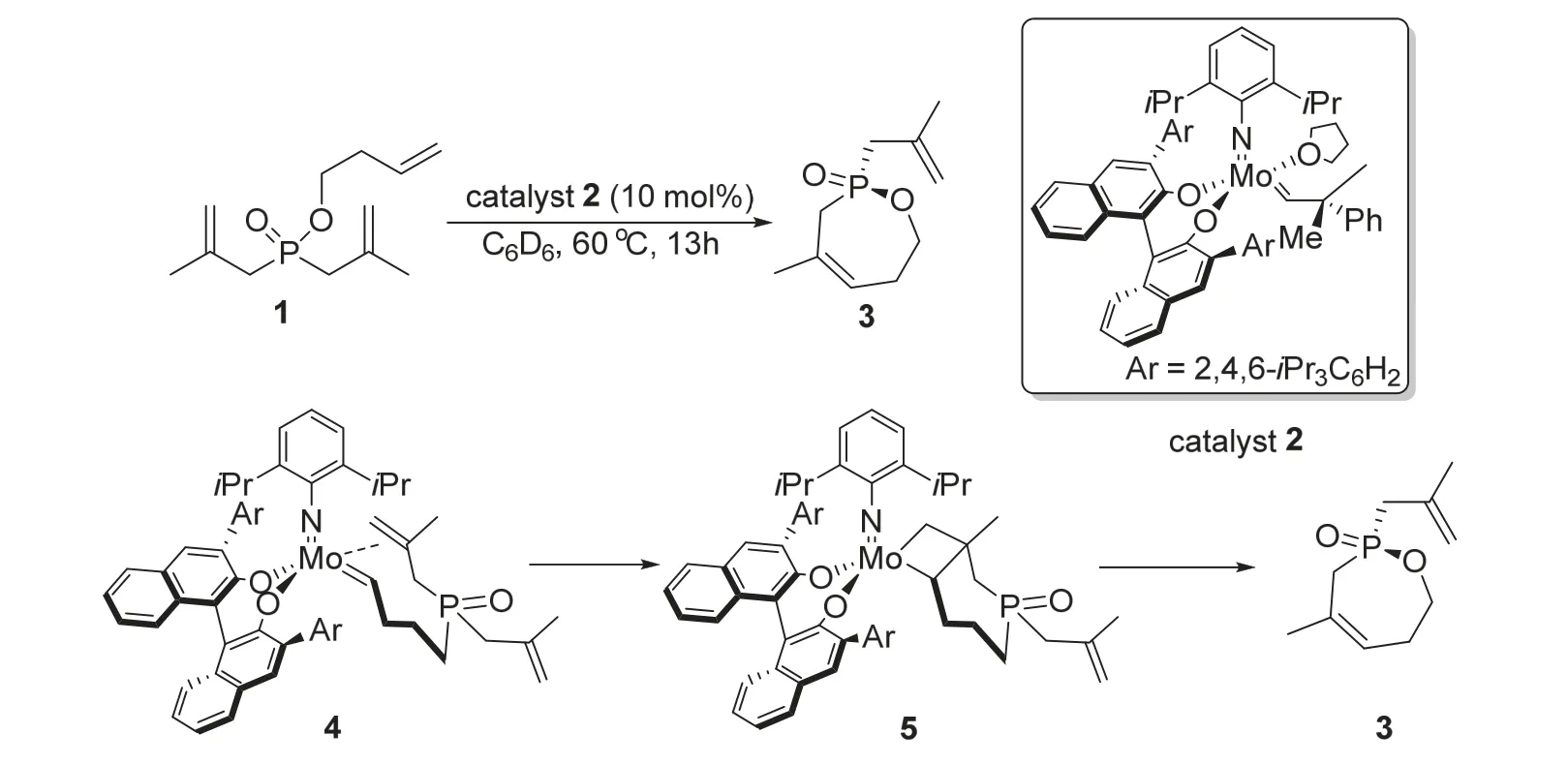
图2 钼催化不对称合成手性磷杂环化合物
2015年,段伟良课题组[9]报道了利用钯催化对映选择性C—H键芳基化反应合成手性磷杂环的方法。反应以二芳基次膦酰胺6为底物,筛选了多种磷配体及其他反应条件后,确定了底物6在醋酸钯和配体7的催化下,加入磷酸钾和三甲基乙酸作为反应助剂,80 °C下在甲苯中反应12-72 h,能以最高94%的收率和90%的ee值得到手性六元磷杂环化合物8 (图3)。这种磷杂环化合物在具有高底物适用性的同时,还可以在亲核试剂的作用下发生P—N键的裂解,化合物9能够在烷基锂试剂作用下开环得到联苯基手性磷化合物10。该反应的优点在于所得的手性磷杂环产物可以灵活地开环,从而得到多种具有不对称催化潜力的手性磷骨架化合物。
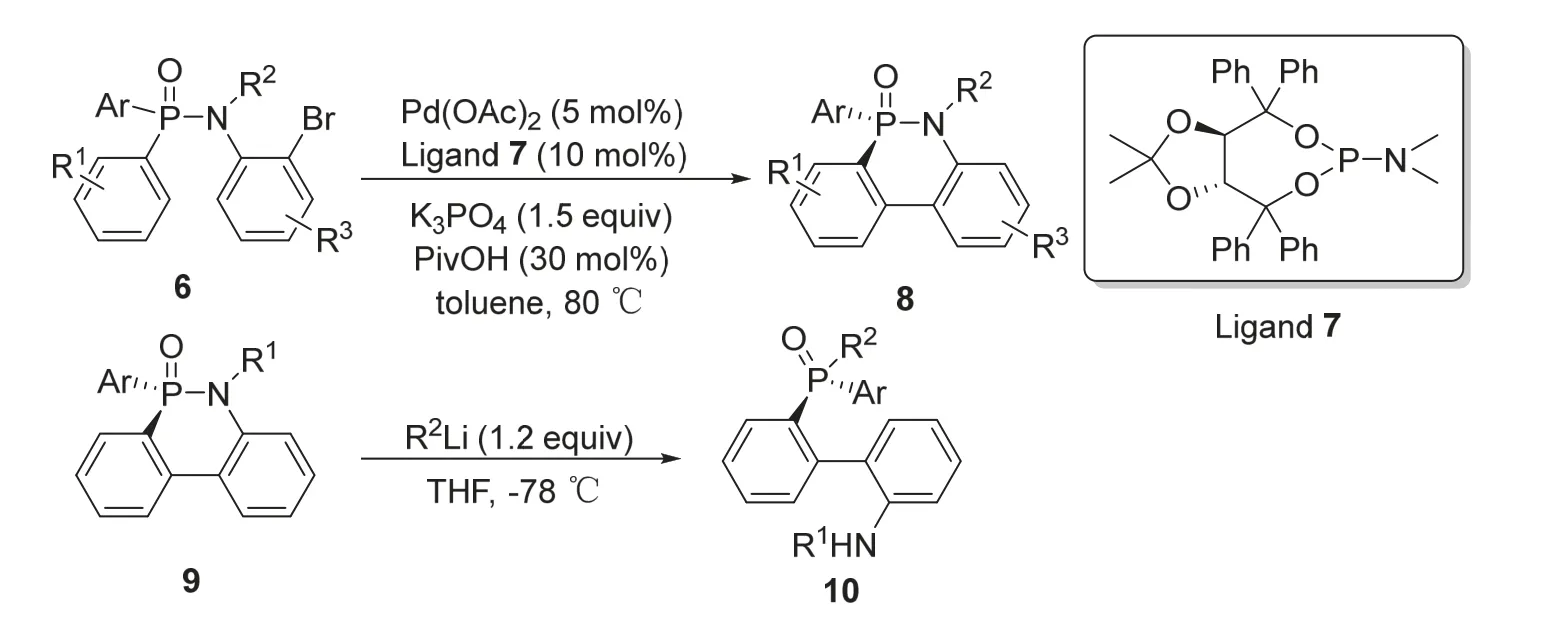
图3 钯催化不对称合成手性磷杂环化合物
2016年,Cramer课题组[10]报道了一种铑催化不对称C—H键活化合成手性环状δ-次膦酰胺的方法。非手性的次膦酰胺底物11和炔烃化合物12在铑催化剂13和过氧化二苯甲酰的作用下,以碳酸银和碳酸钾分别作为氧化剂和碱,在叔丁醇中90 °C反应16 h,能以最高85%的收率和90%的ee值得到手性的环状δ-次膦酰胺产物14 (图4)。该反应的机理是铑催化剂与过氧二苯甲酰作用后与底物15反应,得到中间体16。由于配体的位阻效应带来的影响,铑催化剂部分会靠近底物中特定的苯环从而实现对映选择性的C—H键活化得到中间体17,最后与二苯乙炔反应得到手性环状δ-次膦酰胺18。
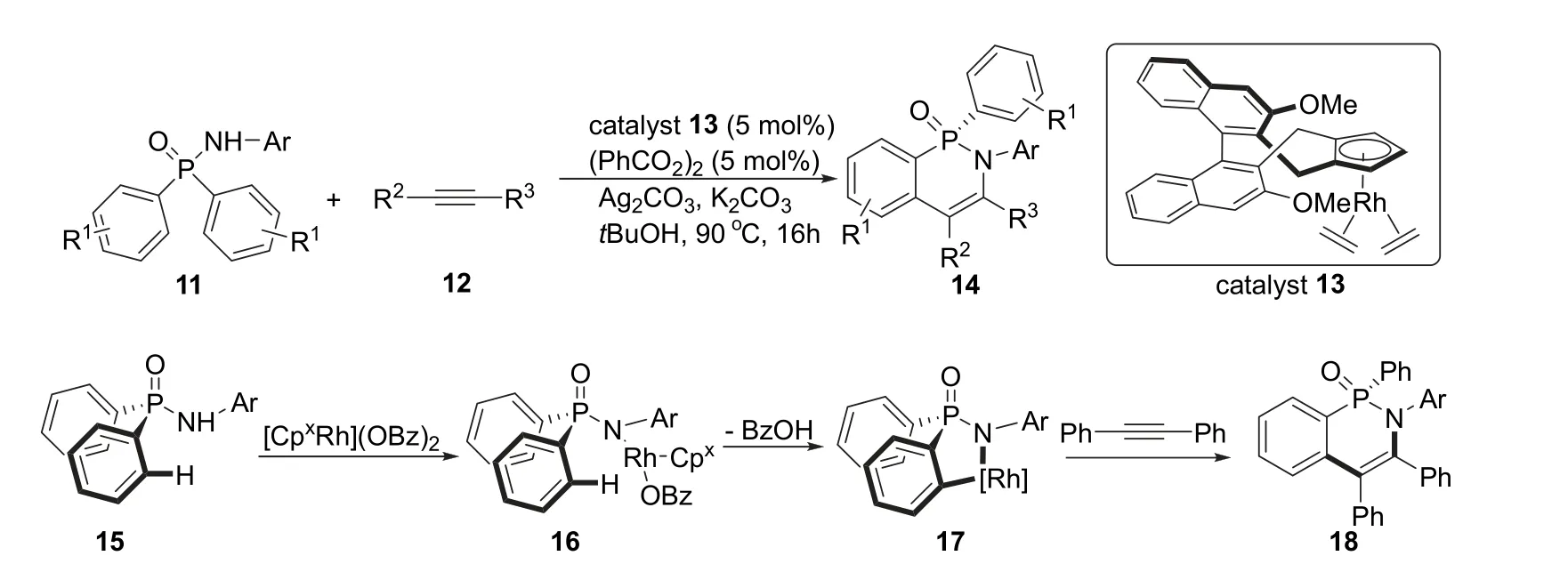
图4 铑催化不对称合成手性磷杂环化合物
2018年,资伟伟课题组[11]报道了一种金催化的对映选择性加氢醚化生成手性磷杂环化合物的方法。反应以双酚19为底物,在氯化亚金、配体20和双氟磺酰亚胺银的催化体系作用下,在甲苯中-20 °C下反应,能以大于95%的收率和最高99%的ee值得到手性磷杂环产物21 (图5)。之后,作者将19的一个苯酚取代基换成炔基,发现在类似的条件下,新的底物22也能在类似的条件下,以仅略微降低的收率和ee值得到手性磷杂环产物23。这种金催化的不对称加氢醚化的方法,实现了手性产物21和23的高对映选择性合成,该反应的底物适用性广,为手性磷杂环化合物的高效提供了新的方法。
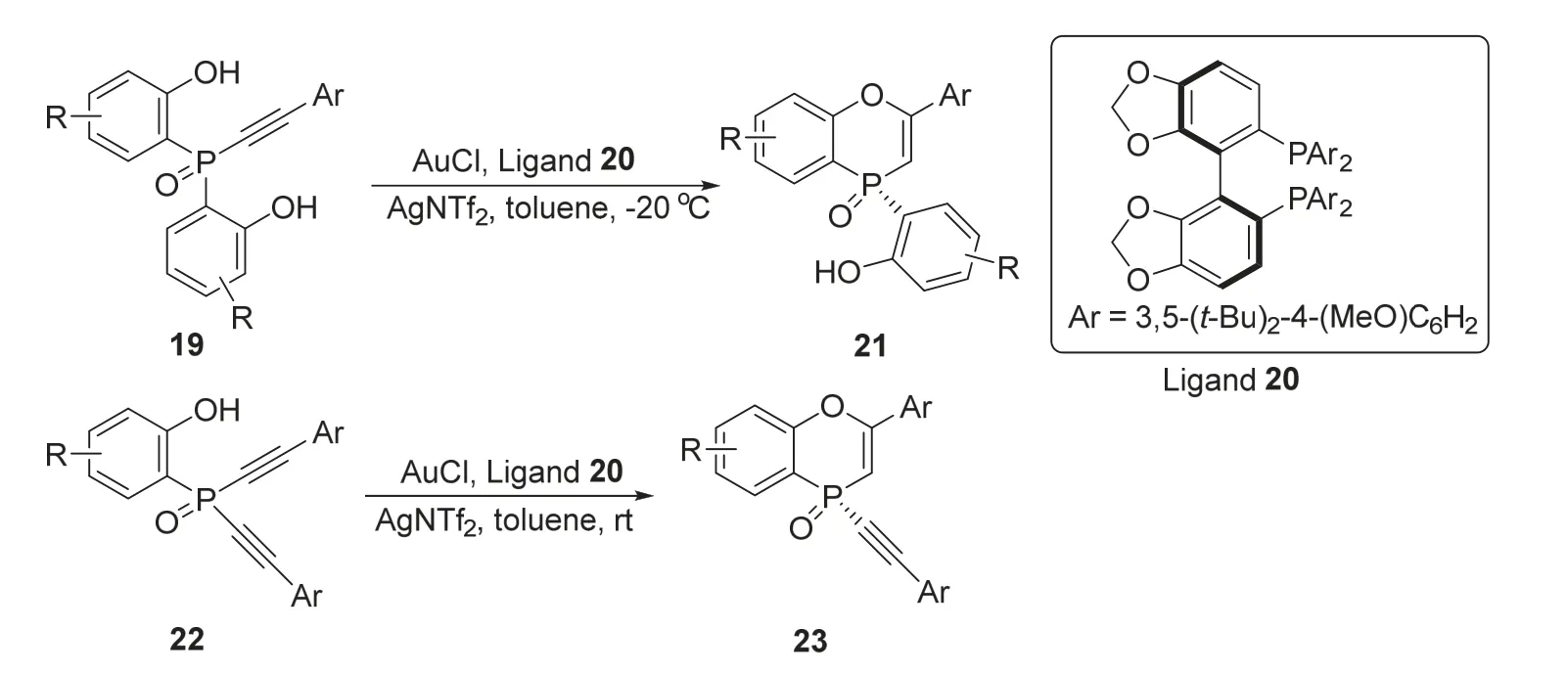
图5 金催化不对称合成手性磷杂环化合物
2022年,史炳锋课题组[12]报道了一种钴催化的不对称C—H键活化合成手性磷杂环化合物的方法。作者使用非手性的次膦酰胺24和炔类化合物25作为底物,在钴金属和配体26的催化作用下,50 °C与四水合乙酸锰等反应助剂作用后能以最高99%的收率和大于99%的ee值得到手性的环状次膦酰胺产物27 (图6)。该反应的机理如图所示,二价钴盐和配体26配位,原位氧化生成三价钴催化剂28,其与底物24进行配体交换后得到中间体29。29的这种构型主要是因为底物24的Q基团和一个苯环,分别与配体26的苯环和苯酚基团具有π-π堆积相互作用,这种作用区分了24的两个苯环。随后底物25与中间体29发生配体交换得到中间体30。由于炔基上取代基与和磷相连的苯基的空间位阻作用,30发生迁移插入反应得到中间体31,最终经过还原消除反应生成目标产物27并释放出钴催化剂32,32在四水合乙酸锰的氧化作用下,从一价回到三价生成28完成催化循环。
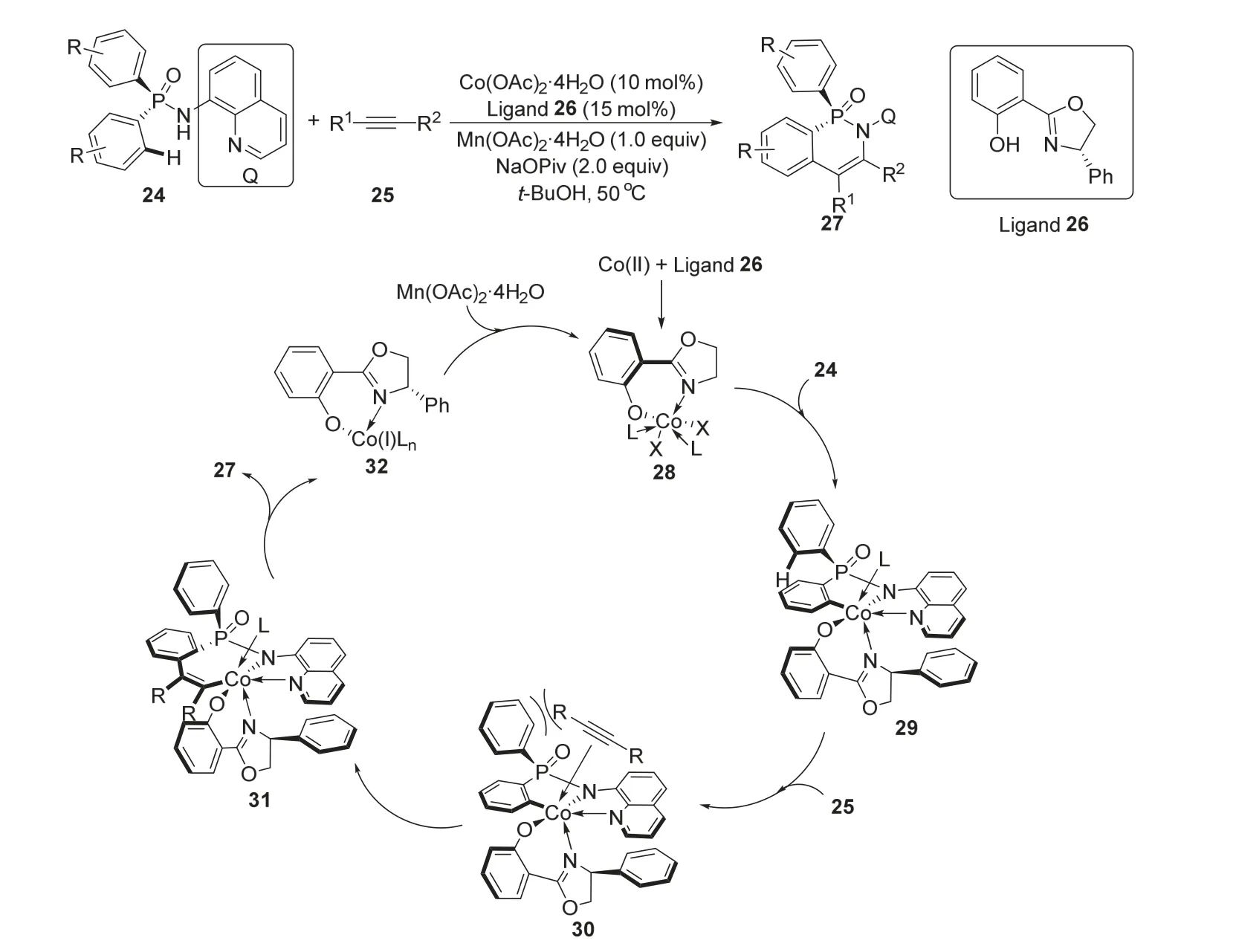
图6 钴催化不对称合成手性磷杂环化合物
2 利用有机小分子促进不对称合成手性磷杂环化合物
2001年,Kobayashi课题组[13]报道了一种以磷杂环戊烷衍生物为底物的不对称羧基化反应。该反应将苯基磷杂环戊烷甲硼烷络合物33与当量的鹰爪豆碱34及仲丁基锂在-78 °C条件下反应3 h后,加入干冰,即可得到脯氨酸型的手性磷杂环产物35 (图7)。该反应的反应收率、dr值和ee值最高可分别达到79%、36 : 1和90%。该反应的创新性在于底物33可以由1,4-二溴丁烷便利地制备,因此该方法提供了一种从简单原料出发制备五元手性磷配体骨架的新思路。
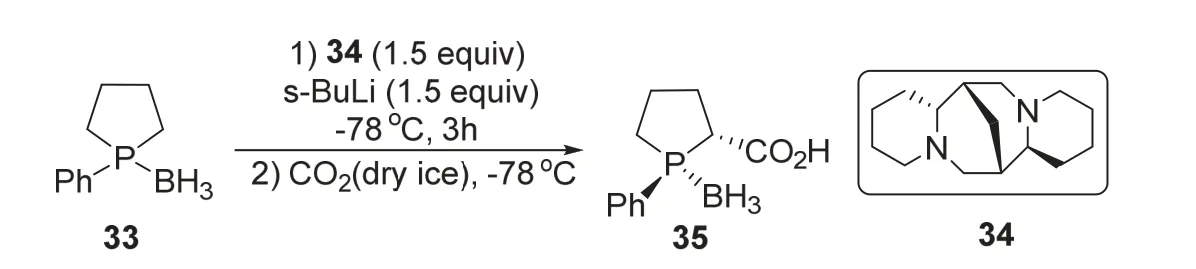
图7 鹰爪豆碱促进不对称合成手性磷杂环化合物
2014年,Johnston课题组[14]报道了一种利用手性有机小分子催化分子内不对称加成反应生成手性磷杂环化合物的方法。该反应以N-烯丙基磷酰胺酸衍生物36为底物,在手性布朗斯特酸37的催化下,与碘代琥珀酰亚胺反应,实现了卤素和磷酰胺酸对碳-碳双键的分子内对映选择性加成,得到含有磷、碳多手性中心的磷杂环产物38 (图8)。该反应最高可以95%的收率,大于20 : 1的dr值和98%的ee值得到目标分子。该反应主要利用磷酰胺酸同时具有的布朗斯特酸性和布朗斯特碱性,使得手性布朗斯特酸37可以利用氢键作用与底物36连接,并利用37中的大基团所产生的位阻效应控制产物中各基团的空间位置,从而实现立体选择性。该方法的创新性在于一步构建了多个手性位点,为手性小分子催化的不对称合成提供了新的思路。
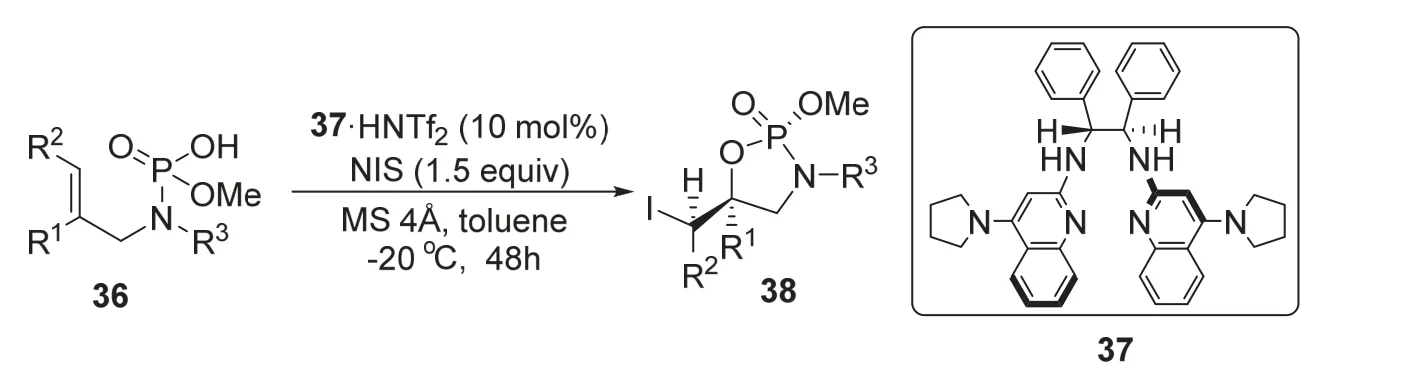
图8 手性布朗斯特酸催化不对称合成手性磷杂环化合物
2021年,Pietrusiewicz课题组[15]报道了一种硫代磷杂环酮一锅法对映选择性合成磷手型烯酮化合物的方法。该反应以1-苯基磷杂环己-4-酮-1-硫化物38为底物,在(S,S)-双-(1-苯基乙基)胺39、氯化锂、正丁基锂、三甲基氯硅烷的作用下,经过不对称去质子化,以76%的ee值得到中间体40。随后在DMSO中与2-碘酰基苯甲酸(IBX)和甲基丙二醇(MPO)反应,将40原位氧化,以49%的收率和73%的ee值得到目标产物41 (图9)。化合物41经过重结晶后ee值可以达到96%。将底物中的硫换成氧后,作者尝试多种催化剂和反应条件,均效果不佳,最高仅得到47%的收率和34%的ee值。该反应的意义在于把以往全碳环酮的对映选择性去质子化反应拓展到了磷杂环酮上,获得了一种新型手性磷杂环骨架分子。该类磷杂环化合物有望发展成为一类新型含磷手性配体,应用于不对称合成领域。
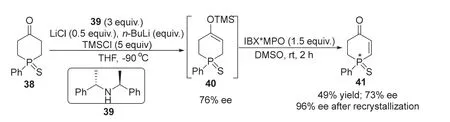
图9 手性胺促进不对称合成手性磷杂环化合物
3 利用磷手性反应底物不对称合成手性磷杂环化合物
2017年,杨尚东课题组[16]报道了一种无金属催化的分子内C—H键活化关环反应,合成兼具轴手性和P手性的磷杂环化合物的方法。作者以手性次膦酰胺42作为反应底物,乙腈为溶剂,在80 °C与碘单质和二乙酸碘苯作用,发生分子内环化和碘代反应并诱导出轴手性,最终以最高78%的收率和大于20 : 1的dr值获得具有轴手性和磷手型的磷杂环产物43 (图10)。反应机理为底物42首先在二乙酸碘苯和碘的作用下生成带有不稳定N—I键的中间体44,随后N—I键均裂得到自由基中间体45。接着N自由基电子被芳基捕获,发生关环反应得到中间体46,进一步在二乙酸碘苯作用下恢复芳环结构得到化合物47。最后化合物47的芳环被游离的碘自由基碘化得到目标产物43。该方法的优点除了温和的反应条件和优秀的立体选择性外,还成功在芳环上实现了碘化,使得反应产物可以进一步发生偶联反应从而定向合成骨架更加复杂的手性磷杂环化合物。
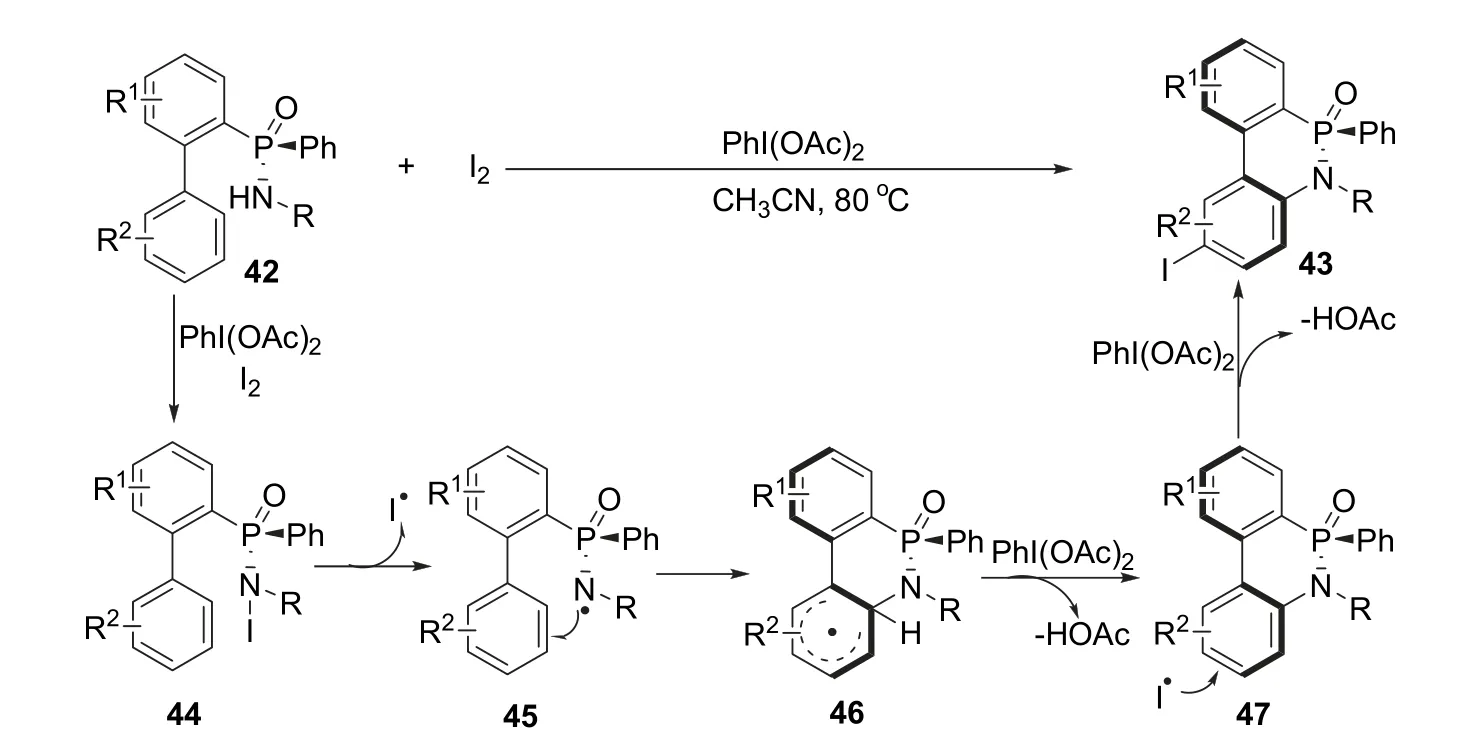
图10 碘促进不对称合成手性磷杂环化合物
2017年,韩福社课题组[17]报道了利用铈促进芳基C—H键活化合成手性磷杂环化合物的方法。该课题组以手性的开链次膦酰胺48为底物,乙腈为溶剂,在2.2当量的硝酸铈铵作用下,50 °C反应20 min,能够以最高93%的收率和97%的ee值得到关环产物49 (图11)。该反应的底物与前述杨尚东课题组所使用的底物类似,但是在N上连有拉电子的五氟苯基后难以再通过N自由基反应机理实现关环反应,因此作者选择了硝酸铈铵作为氧化剂,与底物48作用后得到苯基自由基阳离子中间体50,随后发生关环反应得到中间体51并恢复芳环结构得到目标产物49。该产物还可以在保持ee值的前提下顺利转化为硫代次膦酰胺,随后脱硫得到最高96% ee值的三价磷杂环产物。该方法的创新点在于开发了一种与通过N自由基历程制备手性环状次膦酰胺化合物互补的方法,以苯基自由基阳离子历程实现N上连有拉电子基团的手性环状次膦酰胺化合物的合成,同时也验证了该骨架分子在不对称合成领域具有潜在应用价值。
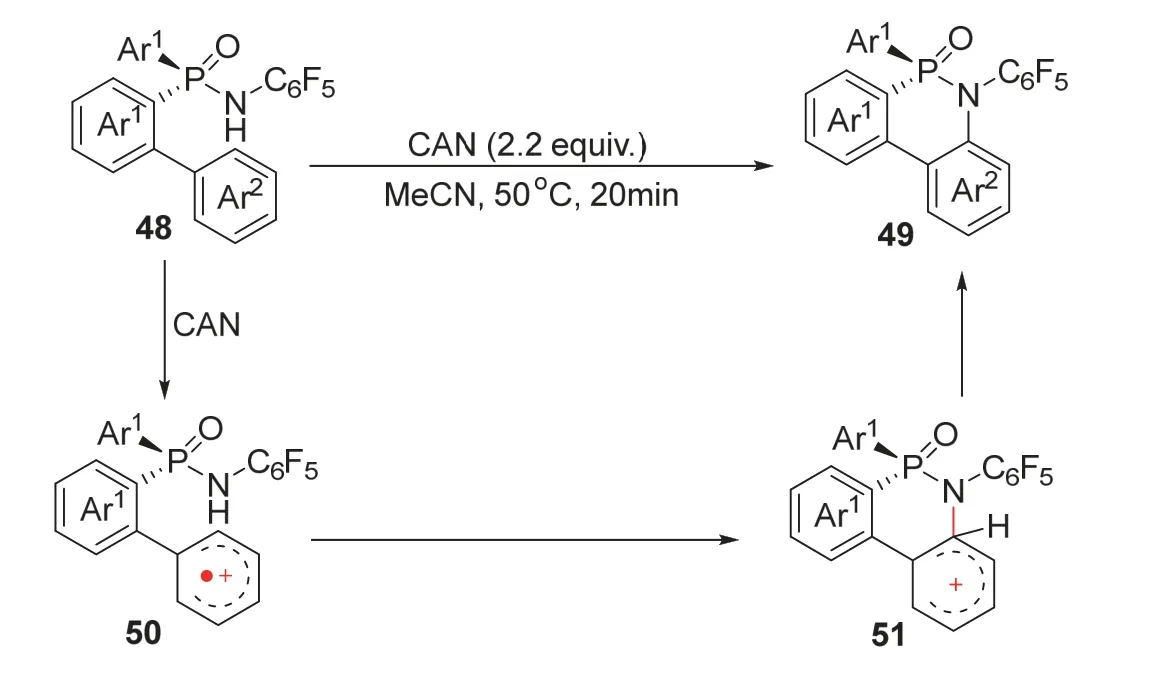
图11 硝酸铈铵促进不对称合成手性磷杂环化合物
2017年,Morandi课题组[18]报道了利用钯催化碳-磷键复分解反应构建手性磷杂环化合物的方法(图12)。作者以(R)-7,7’-双(二苯基膦基)-2,2’,3,3’-四氢-1,1’-螺二茚52为底物,在三(二亚苄基丙酮)二钯和碘苯的催化下,以对二甲苯为溶剂在160 °C下反应24 h,发生碳-磷键复分解反应得到三价膦手性磷杂环中间体53,随后利用过氧化氢将53氧化成在空气中稳定的五价膦手性产物54,反应的总收率为50%而且反应过程中手性始终得到保持(> 99% ee)。在反应过程中碘苯首先与底物52作用得到更易发生氧化加成反应的ArP+Ph3I-中间体(Ar = 螺环芳基,Ph = 苯基),随后ArP+Ph3I-中间体与零价钯(Pd0)依次发生氧化加成、配体交换和还原消除反应实现碳-磷键的构建。该反应的创新点在于探索了一种磷杂环化合物合成的新思路,对碳-磷键复分解反应进行了深入的研究,从而大大拓展了手性磷杂环化合物的合成方法。
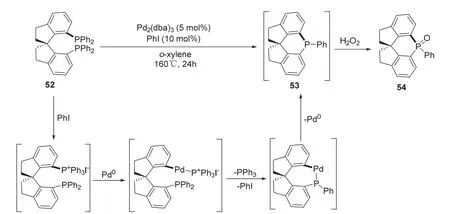
图12 螺环底物不对称合成手性磷杂环化合物
2018年,Ortiz课题组[19]报道了一种先用传统方法制备开链磷手性化合物,随后使其在保持手性的基础上发生分子内成环反应,通过手性诱导策略合成磷杂环化合物的方法。该团队利用Sonogashira反应获得P-苯基-P-(2-炔基苯基)次膦酰胺底物55后,在四丁基氟化铵的作用下,能够在保持原有手性的基础上发生分子内环化反应,并诱导产生高选择性的顺反异构。当R2基团的结构不同时,可以生成56或57两种不同的目标产物(图13),其中产物56的收率和顺反异构比例为88%和92 : 8,产物57的收率和顺反异构比例为95%和91 : 9。该方法的优势在是从简单的磷手性原料出发,在保持了原本手性结构的同时诱导出了新的立体选择性,为手性磷杂环的合成提供了新的方法。
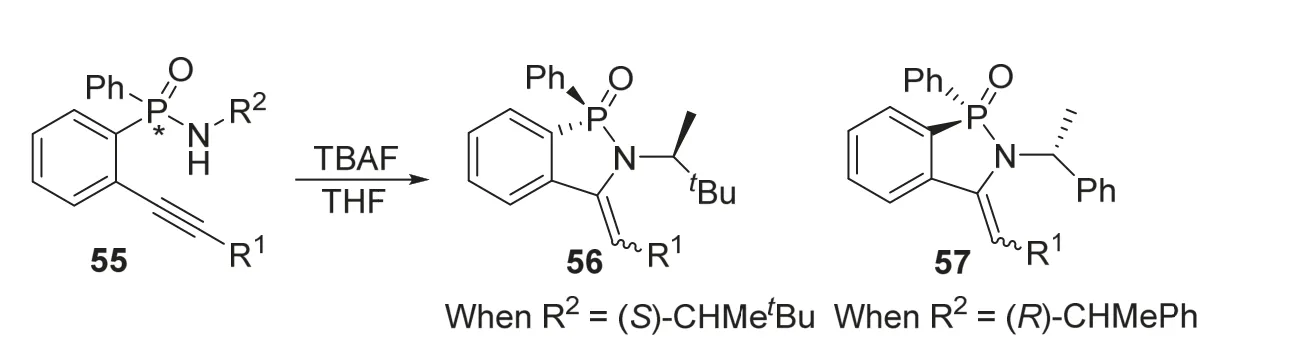
图13 四丁基氟化铵促进不对称合成手性磷杂环化合物
2018年,Glueck课题组[20]报道了利用动态动力学不对称合成(DYKAT)方法合成三元环手性磷杂环化合物的新方法。作者利用手性环氧丙烷58为底物,与膦烷锂盐混合后加入对甲苯磺酰氯发生开环反应得到一对非对映异构体59和60。接着在稀盐酸的作用下脱去对苯甲磺酸关环,通过动态动力学不对称合成得到具有磷-碳双手性的产物61,反应最高收率为74%,最高ee值和dr值均大于99 : 1(图14)。该反应的特点是,一般的膦试剂发生亲核进攻时,磷原子上各基团构型翻转的能垒过大而难以跨越,因此反应过程中会保持构型,但是在该反应的成环过程中,中间产物59和60在分子内亲核取代的过程中均会发生构型翻转,分别形成中间体62和63。这是因为62和63中的碳正离子会被三叔丁基苯基(—Mes*)的超共轭作用所稳定。同时,作者在经过计算后发现,相较于63,62是能量更低、稳定性更高的构型,这使得62转化为64的速度远高于63转化为65的速度,而这两种速度均低于非对映异构体59和60的互相转化的速度。这些原因最终使得两种中间产物不断地向64转化,最终实现了动态动力学不对称合成。
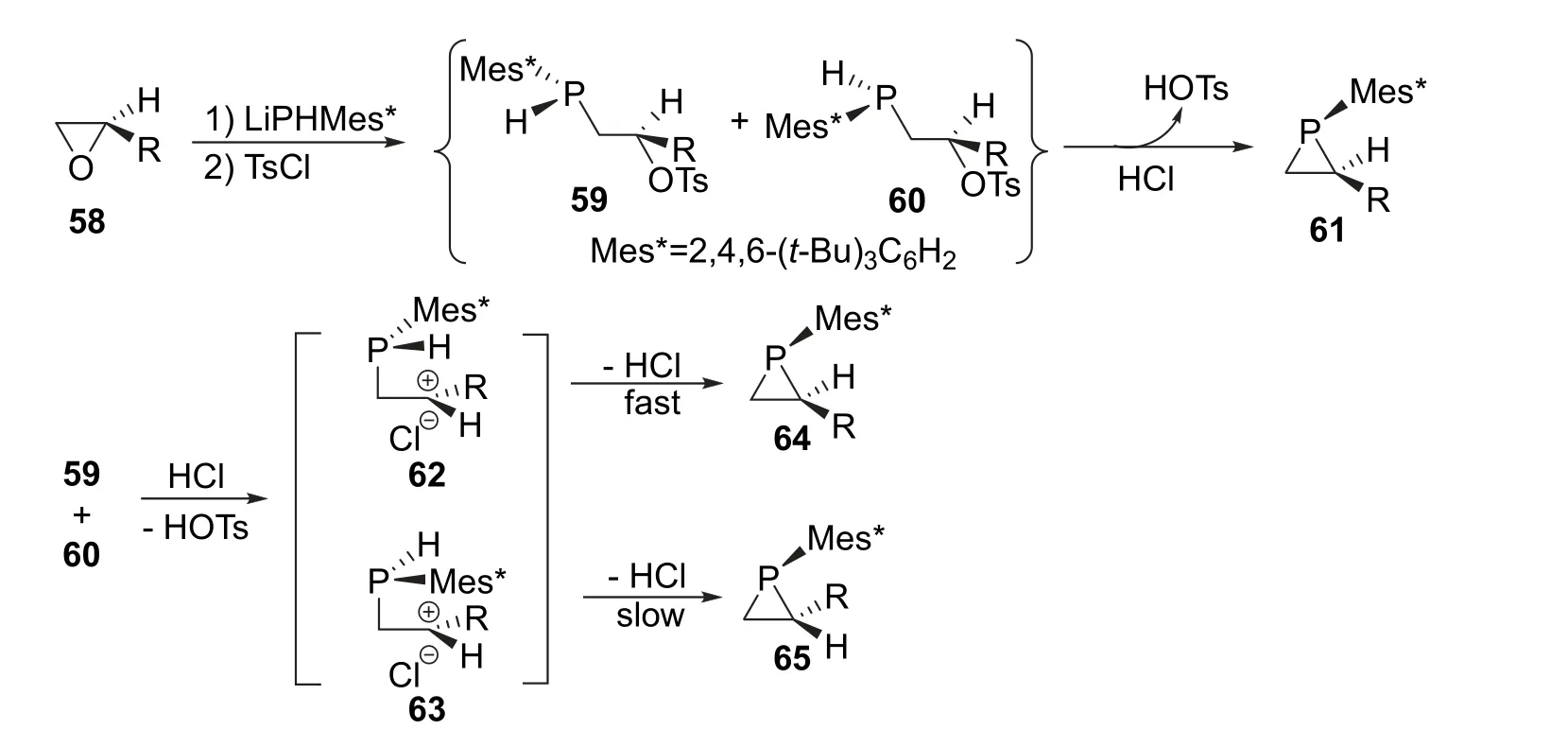
图14 动态动力学拆分不对称合成手性磷杂环化合物
2019年,李强课题组[21]报道了一种联苯基手性磷化合物通过关环反应构建联苯轴手性的方法。作者首先对消旋原料进行重结晶以获得同时具有轴手性和磷手性的底物66和仅具有磷手性的底物67。66在吡啶溶剂中与二氯亚砜作用,能在成环过程中完整地保持手性结构,以78%的收率和大于99 : 1的dr值得到手性八元磷杂环产物68。而仅具备磷手性而没有轴手性的底物67则可以与甲醛反应,以88%和12%的收率得到非对映异构体69和70,随后在氢氧化钾的作用下发生不对称关环反应,最终以大于99%的收率和大于99 : 1的dr值得到具有磷手性和轴手性的七元磷杂环产物71 (图15)。该反应的创新点在于可以利用底物的磷手性在分子内关环的过程中固定轴手性,这种固定作用不但能使原有的轴手性维持稳定,还能定向地控制轴手性的立体选择性合成,从而为合成带有轴手性的膦配体骨架提供了全新思路。
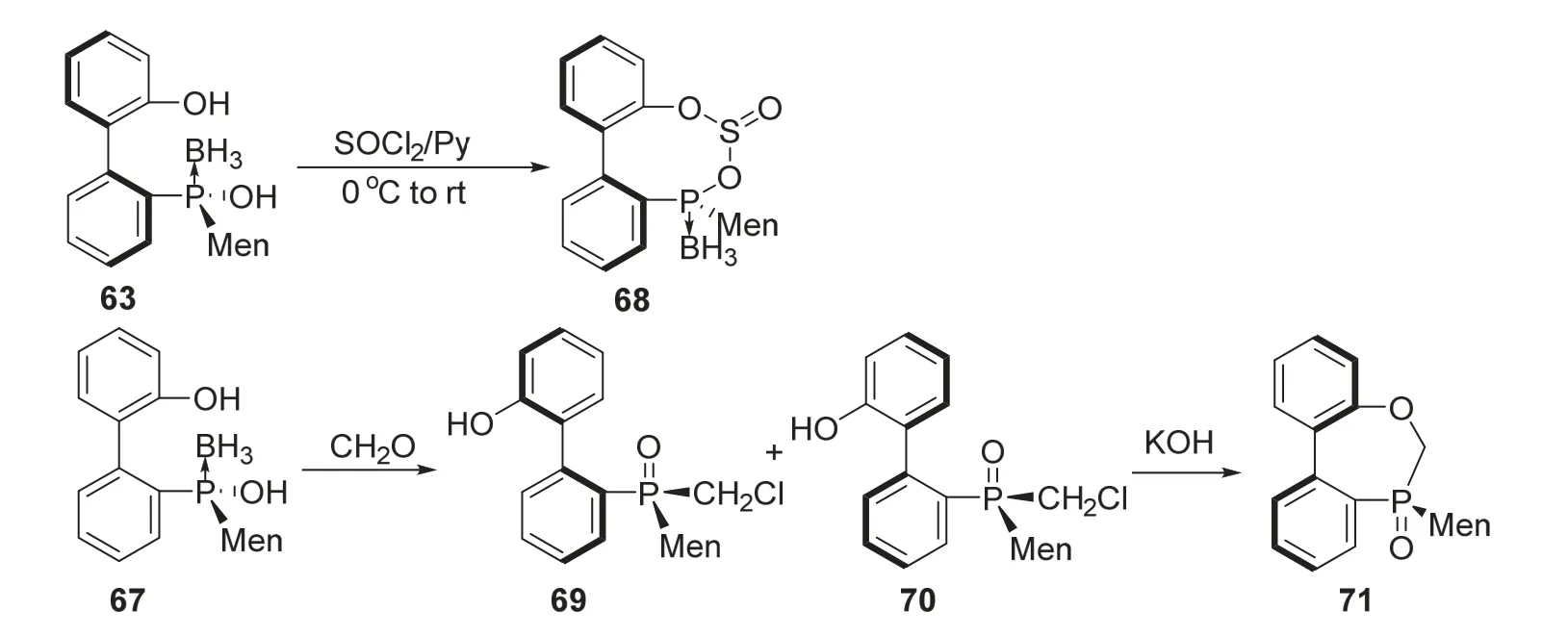
图15 不对称合成具有轴手性的手性磷杂环化合物
2020年,段征课题组[22]报道了利用邻炔基溴苯和手性氧化膦一锅法成环反应合成手性磷杂环戊烷的方法。作者将炔基溴苯衍生物72首先与正丁基锂在-78 °C下反应,随后加入0.5当量的(R)-甲基(噁唑烷酮) (苯基)氧化膦衍生物73,在四氢呋喃中反应并将温度逐渐升至室温即可以最高50%的收率和99%的ee值得到手性磷杂环戊烷产物74。该反应的机理为底物72在正丁基锂作用下得到中间产物75,随后与73偶联得到化合物76,随后正丁基锂作为锂化试剂使其脱质子化得到77,最后发生分子内环化反应生成目标产物74 (图16)。其中正丁基锂的双重角色是需要加入氧化膦底物的两倍当量的原因。该反应的意义在于能够优异地立体选择性合成磷杂环戊烷骨架,为多取代手性膦配体的合成提供了新的合成方法。
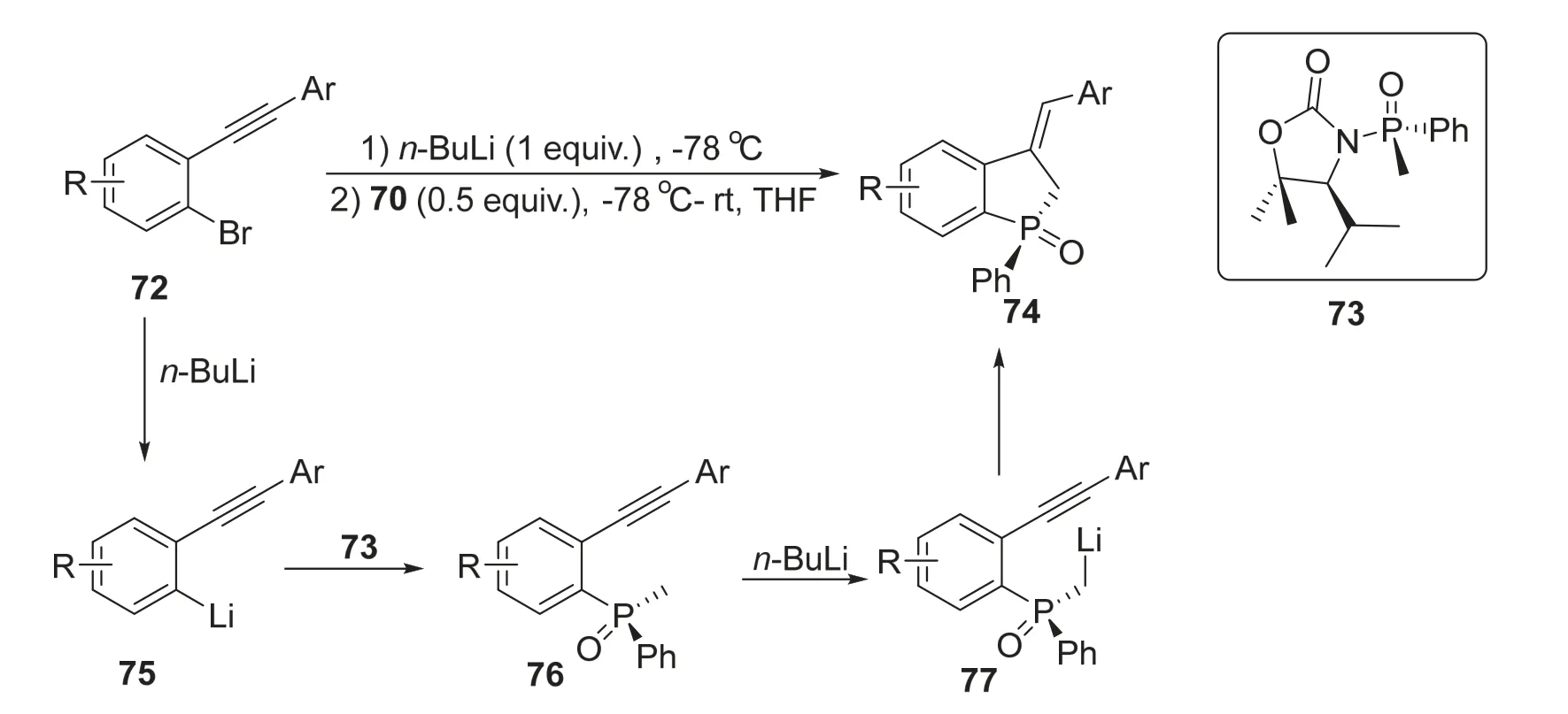
图16 正丁基锂促进不对称合成手性磷杂环化合物
4 手性磷杂环中五价膦到三价膦的转化
目前关于手性磷杂环化合物的报道中以合成五价膦杂环化合物的报道为主,三价膦手性磷杂环化合物的合成方法相对较少。而作为金属配体往往需要三价膦的手性磷杂环化合物[3-6],为了拓宽产物的应用前景,研究者在不对称合成五价膦手性杂环化合物的基础上,通常会将五价膦还原到三价膦化合物。然而在还原过程中不仅需要使原本的磷手性得以保持,还要保证三价膦产物在空气中稳定,这使得该还原反应变得颇为困难,目前仅有少量关于五价膦手性杂环化合物被成功还原到三价膦化合物的报道。
2016年,Cramer课题组[10]报道的利用铑催化不对称C—H键活化反应合成手性环状δ-次膦酰胺的工作中,就将获得的五价膦手性磷杂环底物14,在三氯硅烷的还原下成功以92%的收率和93 : 7的er值得到了三价膦手性磷杂环化合物15 (图17),化合物15在空气中能够稳定存在。
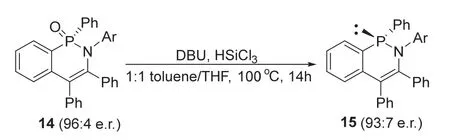
图17 三氯硅烷还原得到三价膦手性磷杂环化合物
在2017年韩福社课题组[17]报道的利用铈促进芳基C—H键活化反应合成手性磷杂环化合物的工作中,同样尝试了五价膦手性杂环化合物的还原反应。他们首先尝试不同的还原剂,例如:三氯硅烷、四氢铝锂、硼氢化钠,但是均无法获得目标产物。最终将次膦酰胺78先在劳森试剂的作用下硫化得到硫代次膦酰胺79,随后在三氟甲磺酸甲酯和三(二甲胺基)膦的共同作用下发生脱硫反应以94%的收率和96%的ee值得到还原产物80 (图18)。
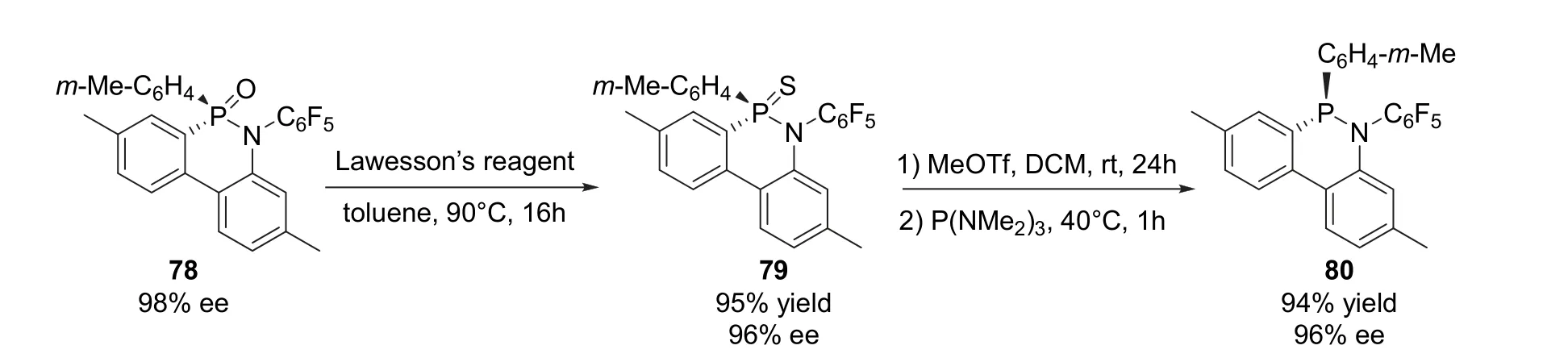
图18 硫代-脱硫方法还原得到三价膦手性磷杂环化合物
2020年,段征课题组[22]报道的利用邻炔基溴苯和手性氧化膦为反应底物合成手性磷杂环戊烷的工作中,对其获得的手性磷杂环戊烷化合物进行了还原反应的尝试。在以三氯硅烷作为还原剂室温搅拌10 min后即得到三价膦的手性磷杂环化合物82,这种结构无法在空气中稳定存在,因此需要原位加入四氢噻吩氯化金作为络合剂,反应最终以95%的收率获得空气中稳定的三价膦-金(I)络合物83(图19)。
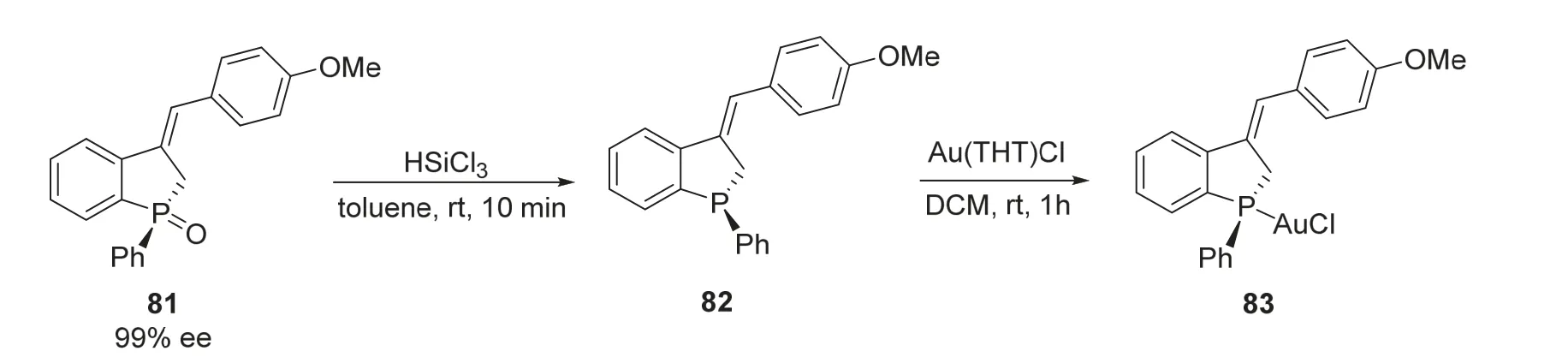
图19 还原-络合方法得到三价膦手性磷杂环化合物
5 结语
手性磷杂环化合物既可以作为手性小分子催化剂直接催化不对称合成反应,也能作为金属配体诱导出手性,在不对称合成领域具有重要应用。在手性磷杂环化合物的合成中,需要兼顾关环与构建磷手性中心两个问题。目前合成手性磷杂环化合物的方法包括金属催化不对称合成、手性小分子促进不对称合成以及磷手性底物环化合成。上述三种合成方法各有优缺点,具体表现如下:(1) 金属催化不对称合成通过金属原子活化底物上的特定官能团促进环化反应进行,同时依靠手性配体与底物的相互作用来构建磷手性中心,但是该方法反应所使用的过渡金属往往比较昂贵而且需要特定构型的手性配体,造成成本较高难以实现大规模生产;(2) 利用手性小分子促进的不对称合成反应构建手性磷杂环化合物,不需要使用贵金属和结构复杂的手性配体,但是仅仅依靠手性小分子催化难以很好地兼顾关环和磷手性构建两个要求,因此该类反应对底物的结构要求较高,反应适用范围不广;(3) 利用含磷手性底物直接环化的方法合成手性磷杂环化合物,无需另外构建新的磷手性中心,仅需解决关环的问题,使这类反应在底物骨架和反应助剂的选择上具有很高的自由度,缺点是反应需要以含磷手性化合物作为底物进行反应。未来该领域的研究重点包括:(1) 发展不对称合成磷杂环化合物的新方法,在合成五价膦手性杂环化合物的基础上,深入研究三价膦手性磷杂环化合物的合成方法;(2) 丰富手性磷杂环化合物的分子骨架结构,拓展手性磷杂环化合物的应用领域。未来随着有机磷化学研究的不断深入,手性磷杂环化合物的合成和应用一定会迎来更大的发展。
猜你喜欢
分子催化(2022年1期)2022-11-02
新作文·中学作文教学研究(2022年4期)2022-08-25
太原科技大学学报(2020年3期)2020-06-22
世界农药(2019年4期)2019-12-30
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
合成化学(2015年2期)2016-01-17
合成化学(2015年9期)2016-01-17
支点(2015年11期)2015-11-16
郑州大学学报(理学版)(2014年3期)2014-03-01
