
高效液相色谱法测定乳香药材中乳香酸多组分含量的方法研究
2024-01-23 13:24豆小卫田彩虹耿娜梁晓莉李惠
陕西中医药大学学报 2024年1期
豆小卫 田彩虹 耿娜 梁晓莉 李惠
(1.陕西冯武臣大药堂有限公司,陕西 咸阳 712000;2.陕西步长制药有限公司,陕西 咸阳 712000)
乳香为橄榄科植物乳香树及同属植物的树干切口中渗出的坚硬胶状树脂。分为索马里乳香和埃塞俄比亚乳香[1]。辛、苦、温,归心、肝、脾经,具有活血行气止痛,消肿生肌之功效。主要用于痛经、经闭、胃痛、心腹诸痛、风湿痹痛;跌打伤痛、痈疽肿痛、肠痈;疮疡肿痛或溃久不收口等[2-7]。在院方和成方制剂中应用广泛[8-10],而且为进口名贵药材。近年来,特别是进口名贵药材的掺假比较普遍[11-15],质量控制尤为重要。在2020年版《中国药典》一部中乳香药材的含量测定为挥发油测定。此方法专属性不强,可能给企业带来经济损失和失信风险。而且现有含量测定文献不多,大多基于挥发油研究,个别仅针对一种成分,不够科学[16-20]。因此,为了解决这一问题,对乳香药材的代表性成分[21-27]进行分析研究,建立代表性成分乳香酸多组分含量测定的高效液相色谱法,为乳香药材质量控制和用药安全提供依据。
1 材料与仪器
1.1仪器 安捷伦1260高效液相色谱仪(UV检测器),UV 2600紫外-可见分光光度计(日本岛津公司),MSA6.6S-OCE百万分之一电子天平(德国赛多利斯公司),KQ-500VDE超声波清洗器(昆山超声仪器有限公司),数显电子恒温水浴锅(天津泰斯特仪器有限公司)。
1.2试剂与试药 对照品α-乳香酸(批号M14GB14667,纯度99.3%)、β-乳香酸(批号M14GB14668,纯度98.4746%)、11-羰基-β-乙酰乳香酸(批号111760-201502,纯度99.3%)均购自中国食品药品检定研究院。甲醇、乙腈为色谱纯(赛默飞世尔科技公司),其余试剂为分析纯(国药集团化学试剂有限公司),3批乳香药材(批号0216321002、0216321003、0216321004)均购自陕西地道药材有限公司。
2 方法
2.1系统色谱条件 采用Agilent1260-C8(150 mm×4.6 mm,5 μm)色谱柱,柱温35 ℃,乙腈-0.1%磷酸为流动相,80∶20(测定α-乳香酸,β-乳香酸;检测波长210 nm),70∶30(测定11-羰基-β-乙酰乳香酸;检测波长250 nm),流速1.0 mL·min-1,进样量10 μL。
2.2溶液制备
2.2.1对照品溶液制备 分别取α-乳香酸、β-乳香酸、11-羰基-β-乙酰乳香酸对照品适量,精密称定,分别加甲醇溶液制成42.34 μg·mL-1、50.81 μg·mL-1、41.15 μg·mL-1的对照溶液。
2.2.2供试品溶液制备 取各批乳香药材适量,分别研细混匀的粉末过4号筛,取0.2 g精密称定,置具塞锥形瓶中,精密加入甲醇50 mL,超声处理30 min,过滤,即得。
2.2.3阴性样品溶液制备 取桃胶适量,按供试品溶液制备方法制备阴性样品溶液。
3 结果
3.1方法学考察
3.1.1专属性试验 分别取混合对照品溶液、供试品溶液和阴性样品溶液,按系统色谱条件分别进样10 μL,结果见图1。α-乳香酸、β-乳香酸、11-羰基-β-乙酰乳香酸对照品与样品相同保留时间峰重合,阴性样品在对照品峰和样品峰相同保留时间无干扰峰。检测峰与相邻色谱峰的分离度大于1.5,理论塔板数大于10000。说明该方法专属性强,系统适用性好。
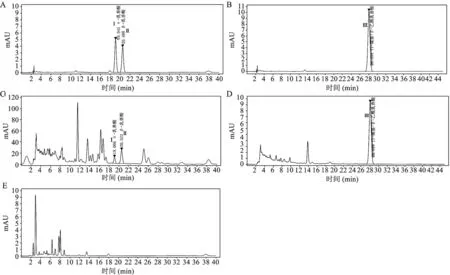
A.对照品1;B.对照品2;C.样品1;D.样品2;E.阴性样品;Ⅰ.α-乳香酸;Ⅱ.β-乳香酸;Ⅲ.11-羰基-β-乙酰乳香酸
3.1.2线性关系考察 分别精密吸取α-乳香酸、β-乳香酸、11-羰基-β-乙酰乳香酸对照品溶液各2、5、10、15、20、25 μL,分别注入高效液相色谱仪中,按确定的色谱条件测定各对照品的峰面积。以对照品进样量为横坐标X,色谱峰面积为纵坐标Y,绘制标准曲线,计算回归方程。结果见表1,可知各成分在各自质量浓度范围内线性关系良好。

表1 各成分线性关系
3.1.3精密度试验 取对照品溶液分别按对应的色谱条件连续进样6次,结果α-乳香酸、β-乳香酸、11-羰基-β-乙酰乳香酸的峰面积RSD(n=6)分别为0.31%、0.29%、0.35%,表明仪器具有良好的精密度。
3.1.4重复性试验 精密称取供试品0.2 g,平行制备6份,按供试品溶液制备方法制备,分别测定含量。结果α-乳香酸平均含量为2.15%,RSD为1.81%;β-乳香酸平均含量为3.94%,RSD为2.47%;11-羰基-β-乙酰乳香酸平均含量为3.33%,RSD为2.26%。表明该方法重复性良好。
3.1.5稳定性试验 取同一供试品溶液(批号:20210102)分别于室温下放置0、2、8、16、24 h按系统色谱条件进样测定、记录峰面积,结果α-乳香酸含量为2.21%,RSD为1.32%,β-乳香酸含量为3.94%,RSD为0.73%,11-羰基-β-乙酰乳香酸含量为3.36%,RSD为1.83%,表明该方法重复性良好。
3.1.6加样回收率试验 取重复性试验项下已知平均含量(α-乳香酸:10.24 mg·g-1、β-乳香酸:35.24 mg·g-1、11-羰基-β-乙酰乳香酸31.32 mg·g-1)的样品0.1 g精密称定,按对照品含量1∶1,精密加入称定的对照品,按供试品溶液方法制备6份,测定含量。计算各成分的平均回收率和RSD。结果α-乳香酸、β-乳香酸、11-羰基-β-乙酰乳香酸的平均回收率(n=6)分别为94.76%,RSD为1.61%;99.40%,RSD为0.84%;100.55%,RSD为1.30%。见表2。
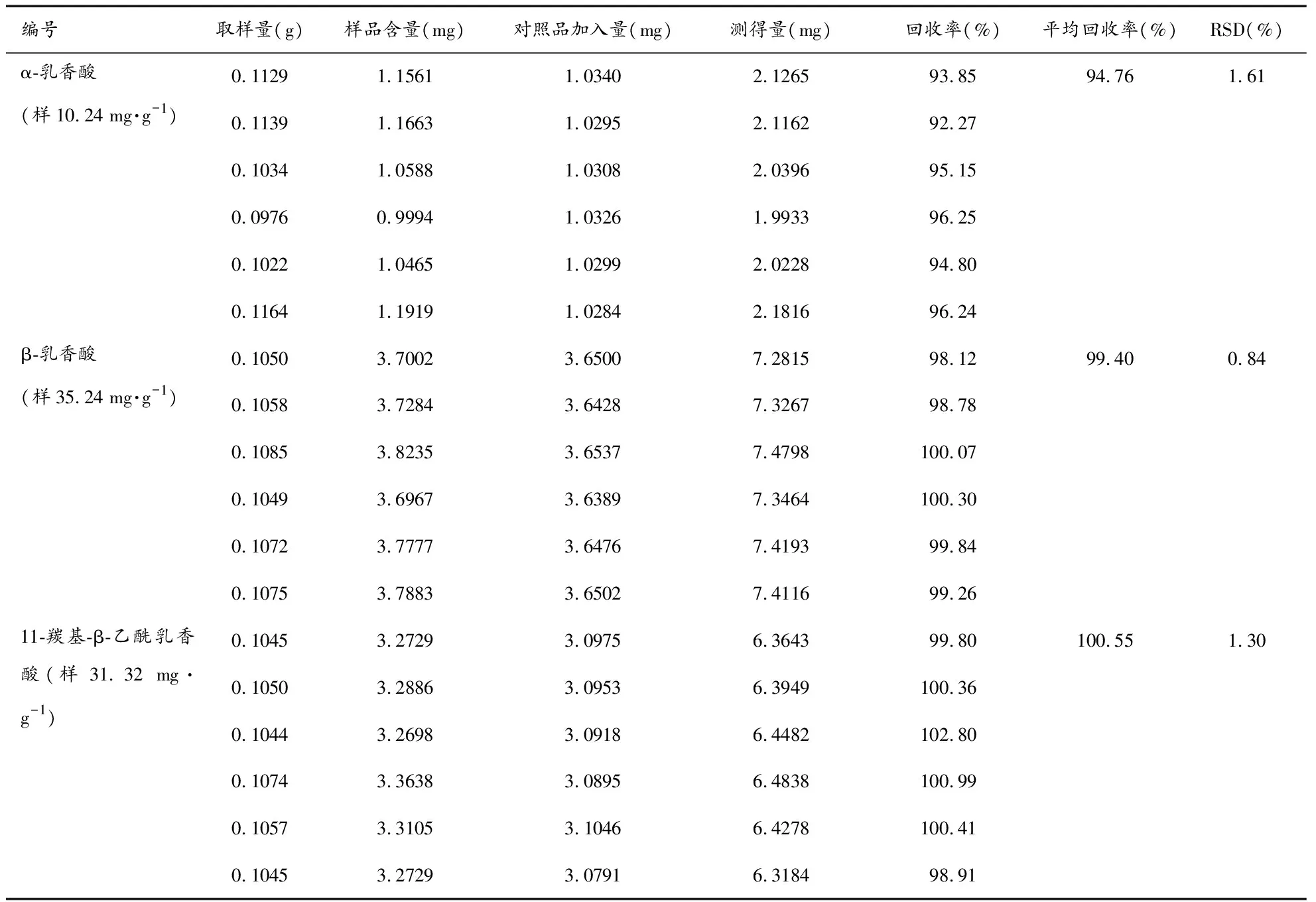
表2 加样回收率测定结果(n=6)
3.2样品测定 取3批乳香药材样品,按供试品溶液方法制品,在系统色谱条件下测定,依据线性方程计算样品中α-乳香酸、β-乳香酸、11-羰基-β-乙酰乳香酸的含量。结果见表3。

表3 样品含量测定结果(mg·g-1)
4 讨论
4.1检测波长的选择 将对照品溶液及供试品溶液分别置于紫外-可见分光光度计中,从200~450 nm波段进行全波段扫描,考虑到末端吸收和排除其他干扰,为获得理想的色谱效果,最终确定α-乳香酸和β-乳香酸的检测波长210 nm;11-羰基-β-乙酰乳香酸的检测波长250 nm。
4.2色谱条件的选择 通过参考文献[28],流动相选择酚酸类成分常用比较理想的相系乙腈:0.1%磷酸溶液。为了简化试验流动相比例选择了20∶80和30∶70两个比例分别测定。此法虽然简便一些,但也可以进一步完善成梯度洗脱。经考察0~15 min(20∶80),16~35 min(30∶70)较为理想。
4.3供试品溶液制备方法 采用加热回流和超声提取方式,设计0.5 h、1 h、2 h分别考察,结果两种方式没有统计学差异。为了简便安全,最终确定供试品溶液的制备方法。
通过试验本文确立了高效液相法对乳香药材中乳香酸多组分的含量测定方法,弥补了现行药典的不足,能客观地评价药材质量。说明该方法简便、可行、专属性强。为乳香药材优劣判定提供科学的依据。
猜你喜欢
长春中医药大学学报(2022年8期)2022-11-21
分子催化(2022年1期)2022-11-02
陶瓷学报(2021年5期)2021-11-22
中成药(2018年9期)2018-10-09
中国民族医药杂志(2016年7期)2016-05-09
云南中医学院学报(2015年2期)2015-07-31
化学反应工程与工艺(2015年1期)2015-04-16
中国塑料(2014年4期)2014-10-17
应用化工(2014年1期)2014-08-16
生物加工过程(2013年1期)2013-03-11
