
Carbyne链力学性能的热依赖性
2024-01-18 08:17贺言罗思林许华慨
广东石油化工学院学报 2023年6期
贺言,罗思林,许华慨
(广东石油化工学院 理学院,广东 茂名 525000)
carbyne作为一种新型的碳同素异形体结构,其由sp杂化形成的一维结构表现出了优异的力学性能和可调控的电子学性质,也为打开纳米结构高性能的电子、光子、自旋电子学设备、灵敏检测设备以及纳米机械器件提供了可能[1,2]。已有的研究表明,carbyne有两种可能的成键结构:由单键和三键交替组成的polyyne和由重复双键组成的cumulene。与金属性质的cumulene相比,由于Peierls畸变驱动键长变化,polyyne表现出的半导体性能将更加稳定[3]。然而,由于carbyne性质对三键和单键交替变化的依赖极强,因而在外界刺激影响下,将驱使内部能量和电子电荷密度重新分配,使得carbyne的力学、热学和电子学性能变得难以捉摸。大量研究表明,衬底、环境和应变引起的三键和单键长度偏移直接影响carbyne的振动和光学性能,甚至引发相变[4,5]。此外,众所周知,热膨胀对于纳米电子和纳米机械器件是至关重要的。纳米材料的热伸长率决定了自身电子学的热力学稳定性和可靠性。Liu[6]和Wong等[7]通过研究发现温度对carbyne的原子特性和总能量有重要影响,这将导致力学和电子性质发生重大变化。虽然人们已经对carbyne的键参数和结构做了大量工作,但明确热效应对单键和三键长度变化的物理机理、确定热膨胀系数,以及了解温度对carbyne力学性能的影响和膨胀的方式依然是基础物理学科中亟待解决的重大问题。
为得到热效应与carbyne的键长和力学性能的关系,基于第一原理计算和Tersoff-Brenner势的理论分析模型,本文对carbyne的热膨胀系数、单键和三键的键长和弹性性能进行了系统的研究。此外,我们还阐明了热引起的波动对杨氏模量和持续长度的物理机制,证实了当温度升高时carbyne将在整个三维空间膨胀,并且carbyne的基本结构为4个碳原子呈单三键排列。
1 理论
考虑到cumulene在较大应变和高温下会因Peierls畸变而不稳定,而具有交替变化键长的polyyne具有更小能量,稳定性更好[8],因此,我们将采用两种不同的方法,第一性原理计算和基于Tersoff-Brenner势的理论框架对比研究polyyne的力学和热性能,其中包括强度、杨氏模量、热容和热膨胀系数等对温度的依赖性。在第一性原理计算方面,采用基于密度泛函理论的VASP (Vienna Ab initio Simulation Package)软件包,计算了48个原子长度的carbyne链电子结构和热容[9],其电子相互作用由广义梯度近似(GGA)[10]方法Perdew-Burke-Ernzerhof (PBE)交换相关泛函描述。基于投影缀加平面波(PAW)方法,电子截断能设置为400 eV。在非周期方向上使用1 nm真空层来消除系统周期相互作用,布里渊区使用1×1×12 Monkhorst-Pack均匀k点网格。另外,在结构优化计算上要求离子实弛豫受力小于0.1 eV/nm,电子步收敛判据为10-6eV,并利用Phonopy程序[11]计算声子色散曲线。最后,利用声子态密度g(ω)可以得到温度T下carbyne链的热容为
(1)
式中:kB和ћ分别为玻尔兹曼常数和约化普朗克常数。
在理论计算方面,考虑carbyne链由交替的单键和三键连接而成,如图1a所示,C—C键在0 K下的势能可以用Tersoff-Brenner势表示为[12]
(2)

图1 C—C单键和三键的能量
(3)
式中:An和Bn为与力场有关的常数,r0和θ0表示碳原子间的距离和平衡态下碳碳键之间的夹角,n为正整数。如果n=4,则上述公式变为Faria公式[13]。此外,考虑carbyne链的应力和弯曲效应的影响,carbyne链在平衡位置附近的能量可表示为
(4)

(5)
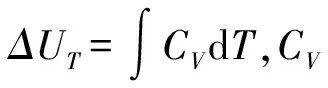
(6)
2 结果和讨论
C—C单键和三键的能量如图1所示。在计算中考虑原子间弹性力的相互作用,应变应小于5%,而且carbyne环引起的弯曲应变对能量的影响可平均计入每个原子中。因此,由公式(4)可得到每两个碳原子的应变能和弯曲能,如图1c和1d所示。另外,根据连续介质力学,carbyne链的拉伸刚度C和弯曲刚度D定义为
(7)
a和R分别为原胞(0.2593 nm)和carbyne环的有效半径。结合公式7,将图1c和图1d用一个多项式拟合,我们可以得到第一性原理计算和理论模型中的拉伸刚度和弯曲刚度分别为C=982 eV/nm,D=0.321 eV·nm和C=1038 eV/nm,D=0.205 eV·nm。这与已有的研究报道基本一致。[6,8,14],而模拟结果与理论预测结果略有差异的主要原因可能是由于计算过程中势能和弹性力的区别导致。在理论计算方面我们采用的Tersoff-Brenner势采用了多项式描述,同时略去了高阶项,因此弹性力学量等力学常数也与模拟计算方面略有差别。此外,由于carbyne链可以被认为是半柔性聚合物,通过计算公式lp=D/(kBT)可以得到室温下相对刚性的持续长度约为6.42 nm。这一结果表明,当carbyne链的长度小于此值时,其将倾向于直链结构以保持较低的弯曲能量;而当大于该长度,其结构完整性将应立即丧失,并产生扭折现象[14]。
为研究carbyne的热性质,我们利用第一性原理通过计算声子色散并代入公式(1)得到了其热容,如图2所示。由图2可知,热容随着温度的升高而增加,这是由于声子在高温下增多的原因所致。此外,与石墨烯的热容相比,carbyne链的热容在低温下比石墨烯要大得多[15],而在高温下两者的热容几乎没有差异。如石墨烯在100 K和300 K时的热容分别为3.9 J/(mol·K)和10.1 J/(mol·K),低于carbyne链的6.9 J/(mol·K)和15.0 J/(mol·K),而在1000 K时这两种结构的热容差为0.4 J/(mol·K)。这一结果表明,carbyne链中的高频声子更容易被激活,而石墨烯在高温下激活的高频声子与carbyne链的高频声子相当。此外,我们预测carbyne链的德拜温度Θ约为510 K,与之前的研究结果一致[6]。表1展示了carbyne链的热学性质。图2的插图展示了能量ΔUT随外部温度增加而增大的关系。
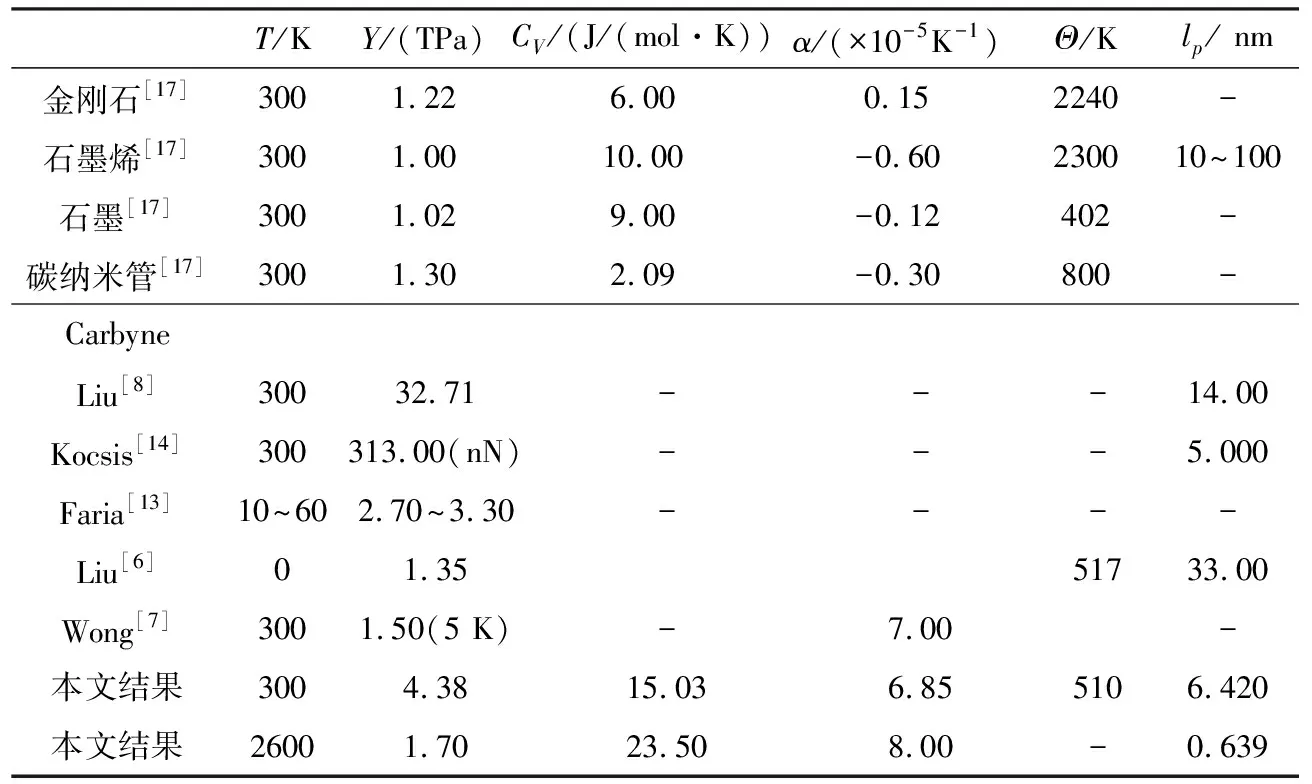
表1 金刚石、石墨烯、石墨、碳纳米管和Carbyne链的力学和热学性能
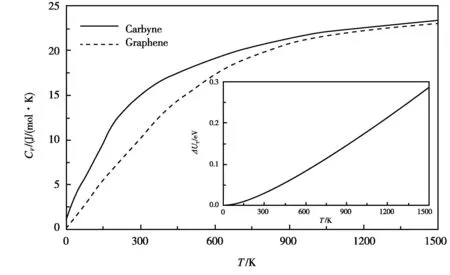
图2 Carbyne链和石墨烯的热熔
图3a展示了carbyne链的原子势和原子键长随温度的变化关系,其单键和三键在0 K下的键长分别为1.348 nm和1.245 nm,并且理论表明最小能量随温度的变化出现显著的漂移,这一结果说明carbyne链自平衡态在温度的影响下将被重新建立,从而导致单键和三键长度的改变,而C—C单键和三键增大的趋势也暗示了carbyne链将随温度的增大而出现正应变。利用公式(6),可以得到carbyne链单键和三键的热膨胀系数随温度的变化关系,由图3可见单键、三键和两者平均值的热膨胀系数,以及利用carbyne链热熵所计算的热膨胀系数。图中表明单键的热膨胀系数略高于三键的,单键和三键热膨胀系数差异的出现是由于弹性性能、力学常数以及键长的不同导致,比如已有的研究[15]证实单键和三键的力学弹性系数在0 K下分别为6.37×103eV/nm2和1.12×104eV/nm2。此外,我们发现carbyne链的热膨胀在整个温度范围内都是正的,并且在低温下随温度的增大而快速上升,当温度超过300 K后则呈缓慢上升趋势,而在常温下其热膨胀约为+6.85×10-5K-1。这一结果与Wong等人(+7×10-5K-1)[7]和Costa工作组(+5×10-5K-1)[16]的结果基本一致,并且与金刚石(+0.1×10-5K-1),石墨烯(-0.3×10-5~-0.9×10-5K-1),石墨(-0.1×10-5K-1),单壁碳纳米管(+0.2×10-5K-1)等相比约大一个数量级[17],如表1所示。
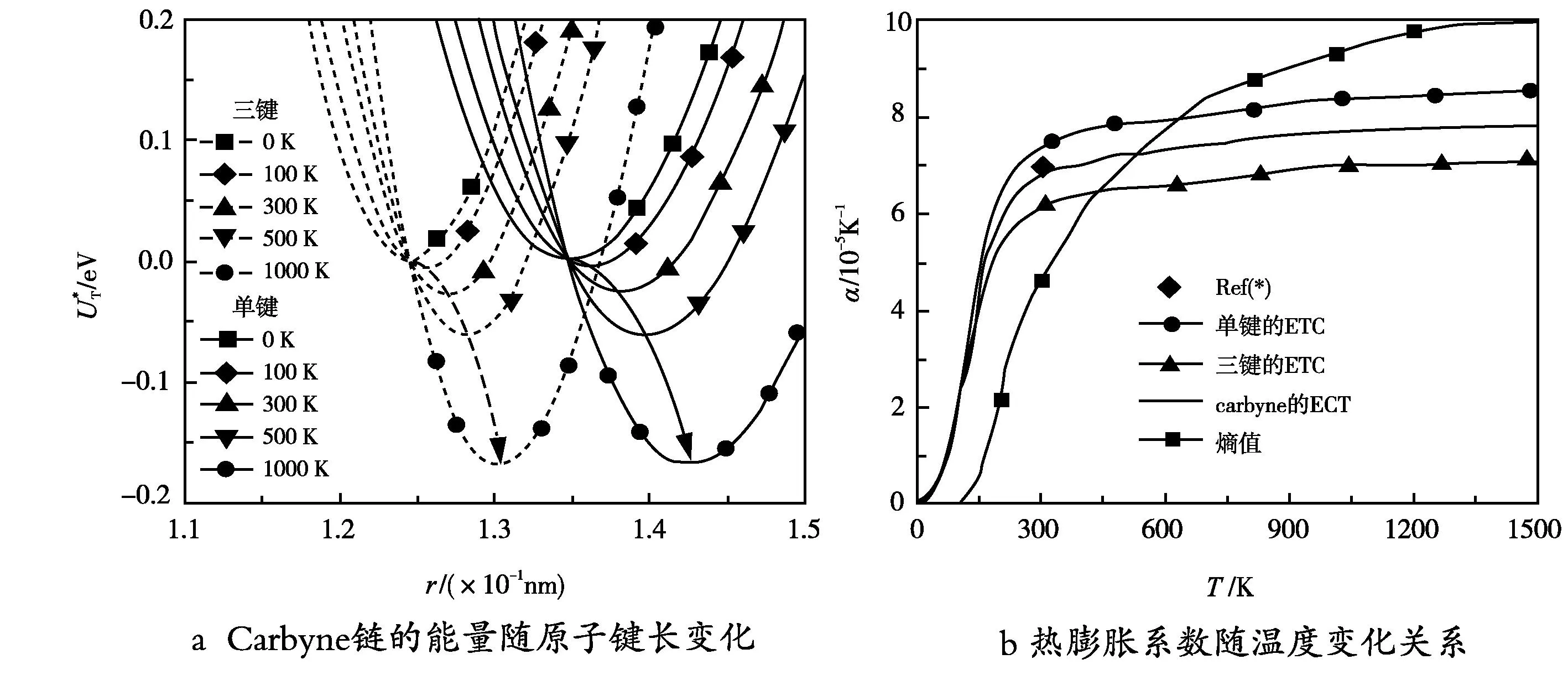
图3 carbyne链的原子势和原子键长随温度的变化
最后,为研究外部温度对力学性能的影响,图4a展示了carbyne链在不同温度下拉伸载荷的应力(σ)-应变(ε)曲线。值得注意的是,carbyne链的断裂应变约为20%,并且该值随温度的升高而减小。因此,我们在0~5%的应力下进行力学弹性常数的研究满足胡克定律要求。如图4a所示,在应变为0%时,carbyne链会出现非零应力且受热波动的影响,非零应力将随温度的增加而增大。这一结果也被之前的工作所报道过[13]。此外,根据杨氏模量的定义Y=dσ/dε,图4b展示了carbyne链杨氏模量随温度的依赖性关系。在0和300 K下,碳链的Y值分别为4.73 TPa和4.38 TPa,其随温度升高而降低的这一趋势可以归结为热效应的增强,并且与模拟结果一致[13,14]。此外,图4b的插图展示了基于Ouyang模型[18]的立方纳米结构应变与杨氏模量的变化关系,Y∝(1+εT)-3。我们的预测与他们的预测相当,这一结果也说明随着温度的升高,carbyne将在整个三维空间内膨胀,而不是只在键长方向膨胀。另外,根据D∝Y[8]的关系,可以得到carbyne链的温度依赖下的持续长度。我们发现当T=2600 K(形成carbyne链所需的温度)时。这一结果略大于carbyne链4个原子键长长度0.591 nm,而小于5个原子键长长度0.745 nm。这一结果表明在高温下合成carbyne链时,其将以4个碳原子为基本单位,这与已有的实验测量结果非常吻合[19]。
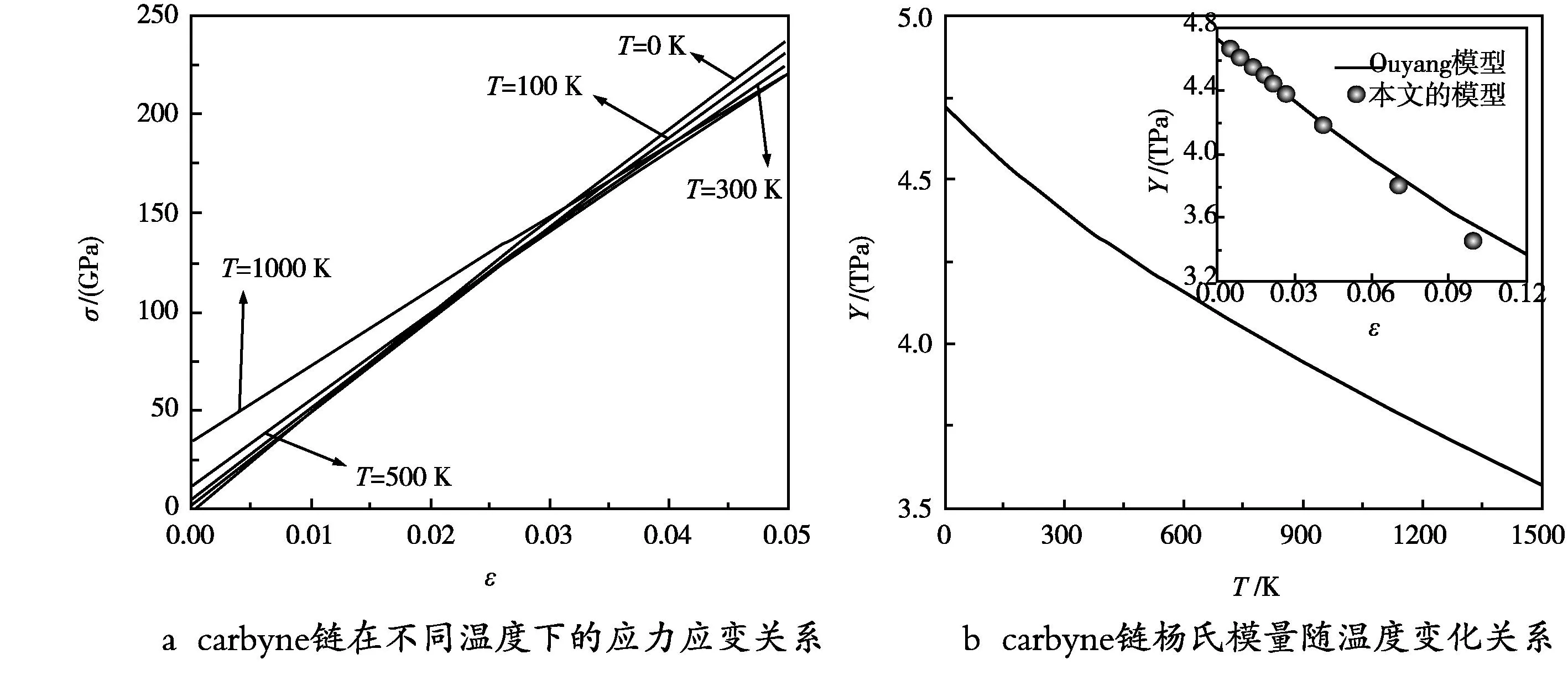
图4 温度对carbyne链的力学性能影响
3 结语
通过第一性原理计算和基于Tersoff-Brenner势的理论分析模型,研究了carbyne链在温度影响下的键参数对其力学性能和热性能的影响。研究表明,carbyne链单键的变化比三键的变化稍大,而且其在整个温度范围内的热膨胀系数都为正。此外,我们的理论预测,随着温度从0升高到1500 K,carbyne链的杨氏模量将由4.73 TPa下降到3.57 TPa,而carbyne链的弹性性质和持续长度都与温度呈依赖关系,这也表明carbyne链是在整个三维空间中膨胀,并且其在高温下是以4个碳原子为基本单位。理论预测与实验测量和模拟结果吻合较好,这也为研究carbyne链的力学和热性质提供了可行的方法。
猜你喜欢
河南科技(2023年10期)2023-06-07
汽车实用技术(2022年5期)2022-04-02
今日农业(2021年7期)2021-07-28
舰船科学技术(2021年12期)2021-03-29
中国测试(2018年10期)2018-11-17
物理实验(2017年2期)2017-03-21
三峡大学学报(自然科学版)(2017年1期)2017-03-20
贺州学院学报(2015年1期)2015-02-28
长江大学学报(自科版)(2014年1期)2014-03-20
中山大学学报(自然科学版)(中英文)(2013年4期)2013-04-24
