
HPLC与UPLC-MS/MS测定FAPAS液体维生素补充剂能力验证样品中的维生素B1
2024-01-16 03:19韦春梦江秋霞
食品与药品 2023年6期
杨 黎,文 丽,韦春梦,江秋霞,刘 星
(1.广西-东盟食品检验检测中心,广西 南宁 530025;2.广西壮族自治区食品药品检验所,广西 南宁 530021)
维生素B1又称硫胺素,具有维持人体正常糖代谢的作用,是糖类代谢酶的组成成分[1],在人体内不能合成,需通过外界间接获取或直接补充[2-3],常作为添加剂制成保健食品。为验证实验室检测保健食品中维生素B1的能力,我们参加了此次英国FAPAS(Food Analysis Performance Assessment Scheme)分析实验室能力验证工作。
维生素B1的检测技术报道较多,主要包括紫外-可见分光光度法[4-5]、高效液相色谱(HPLC)-紫外检测法[6-8]、HPLC-荧光检测法[9]、HPLC-质谱(MS)联用方法[10-11]等。本研究试验对象是FAPAS国际能力验证样品,不指定检验方法。目前国内检测维生素B1的标准主要有GB/T 5009.197-2003《保健食品中盐酸硫胺素、盐酸吡哆醇、烟酸、烟酰胺和咖啡因的测定》[6],GB 5009.84-2016《食品安全国家标准 食品中维生素B1的测定》[9]。基于样品分析国标检测方法,GB/T 5009.197-2003直接用甲醇-水-磷酸混合溶液稀释样品后进行检测,无水解和酶解过程,如样品中含结合态维生素B1有可能提取不完全;GB 5009.84-2016第一法HPLC法需衍生后用荧光检测器进行检测,实验过程长且衍生化合物稳定时间短,当有其他荧光物质干扰时可能影响检测准确性;而质谱检测器在灵敏性、准确度和选择性等方面具有明显优势。综上,在国标的基础上优化了前处理方法,同时采用HPLC-紫外检测和超高效液相色谱-质谱/质谱(UPLC-MS/MS)两种方法进行检测,并比较结果。
1 仪器与材料
1.1 仪器
Waters 2890高效液相色谱仪(带DAD检测器,美国Waters公司);Waters H-CLASS/XEVO TQD三重四极杆液质联用仪(美国Waters公司);X3R高速冷冻离心机(美国Thermo Fisher公司);Milli-Q超纯水机(美国密理博公司);Mettler Toledo XS205DU电子天平(十万分之一,瑞士梅特勒-托利多公司);S300H超声波清洗器(德国Elmasonic公司)。
1.2 材料
液体维生素补充剂样品(由FAPAS实验室提供),样品冷藏存放,测试前按作业指导书放置到常温并充分摇匀。维生素B1标准物质(纯度:98.6 %,CAS:67-03-8,BePure公司,使用前105 ℃干燥4 h);硫酸月桂酯钠,正丁醇,甲醇,乙腈(色谱纯,德国默克公司);铁氰化钾,氢氧化钠,盐酸,乙酸钠,冰乙酸(分析纯,广东光华);木瓜蛋白酶(德国默克公司,酶活力≥1500 U/mg);淀粉酶(德国默克公司,酶活力≥3700 U/g);超纯水(电阻率:18.2 MΩ·cm)。
2 方法
2.1 HPLC
2.1.1 色谱条件 色谱柱:Atlantis T3色谱柱(250 mm×4.6 mm,5 μm);流动相:1 %硫酸月桂酯钠(含0.1 %磷酸)溶液:乙腈=58:42;流速1.0 ml/min;柱温:30 ℃;检测波长:260 nm;进样量:10 μl;等度洗脱。
2.1.2 标准溶液的配制 精密称取25 mg维生素B1标准物质,置于25 ml量瓶中,加水溶解,定容,摇匀,配制质量浓度为1 mg/ml的标准储备溶液。然后以甲醇-水-磷酸(100:400:0.5)为溶剂,配制维生素B1质量浓度分别为0.2,0.5,1.0,2.0,5.0,10.0 μg/ml的标准系列工作溶液。
2.1.3 供试液的制备 称取1.00 g摇匀的试样,置于100 ml具塞三角瓶中,加入0.1 mol/L盐酸溶液60 ml,充分摇匀,塞上软质塞子,高压灭菌锅中121 ℃保持30 min进行水解,水解结束,冷却至40 ℃以下,取出,轻摇数次;然后用2.0 mol/L乙酸钠溶液调节pH至4.0,加入混合酶溶液2.0 ml,摇匀,置于培养箱中37 ℃过夜酶解(酶解时间16 h)。将酶解液全部转移至100 ml量瓶中,用水定容至刻度,摇匀,离心,0.45 μm滤膜过滤,待进样。
2.1.4 加标回收试验 称取1.00 g样品,加入一定量的维生素B1标准溶液,按2.1.3项下方法处理,测定回收率,计算相对标准偏差(RSD)。
2.2 UPLC-MS/MS
2.2.1 色谱条件 色谱柱为ACQUITY UPLC HSS T3色谱柱(100 mm×2.1 mm,1.8 μm),流动相:A为0.2 %甲酸水溶液,B为甲醇;流速:0.3 ml/min;柱温:30 ℃;梯度洗脱程序:0~1 min,100 %A,1~3 min,100 %A~5 %A,3~5 min,5 %A~50 %A,5~6 min,50 %A,6~7 min,50 %A~100 %A,7~11 min,100 %A;进样量:4 μl。
2.2.2 质谱条件 采用ESI源;正离子模式,多反应监测(MRM);毛细管电压:1.2 kV,离子源温度:150 ℃,脱溶剂温度:500 ℃,脱溶剂气流速:1000 L/h,锥孔气流速:150 L/h,碰撞气(氩气)流速:0.15 ml/min。
2.2.3 标准溶液的配制 精密称取25 mg维生素B1标准物质,置于25 ml量瓶中,加0.01 mol/L盐酸溶液溶解并定容,摇匀,配制质量浓度为1.0 mg/ml的标准储备溶液。准确移取1.00 ml标准储备液,用水稀释并定容至100 ml,摇匀,配制质量浓度为10.0 μg/ml的标准中间液,临用现配。然后吸取一定量的标准中间液,配制维生素B1质量浓度为10,50,75,100,200 ng/ml的标准系列工作溶液。
2.2.4 供试品溶液的制备 精密吸取2.1.3项下供试品溶液2.0 ml至10 ml量瓶,加水定容,0.22 μm滤膜过滤,待进样。
3 结果与讨论
3.1 HPLC流动相的优化
首先参照标准GB/T 5009.197-2003[6],使用流动相硫酸月桂酯钠溶液(5 g→530 ml)-乙腈-磷酸(530:470:1),结果采用此流动相,维生素B1色谱峰峰展宽且吸收响应低;更换流动相比例,流动相改为1 %硫酸月桂酯钠(含0.1 %磷酸)溶液-乙腈(58:42),增加水相比例后的色谱峰峰形对称,响应值显著提高,且目标化合物附近没有干扰峰,见图1。

图1 标准溶液和供试品溶液色谱图
3.2 UPLC-MS/MS质谱条件优化
在ESI正、负离子模式下,对维生素B1标准溶液(浓度100 ng/ml)进行母离子扫描,维生素B1在正离子模式下质谱信号强,确定正离子模式扫描;然后在子离子扫描模式下,通过施加一定碰撞能量,使维生素B1母离子碎裂产生子离子,获取子离子信息,并对毛细管电压、离子源温度、脱溶剂温度、脱溶剂气流速、锥孔气流速、碰撞气(氩气)流速、锥孔电压、碰撞能量等质谱参数进行了优化,以强度最大的子离子作为定量离子,以强度稍小的子离子作为定性离子。优化后的质谱检测参数见表1。
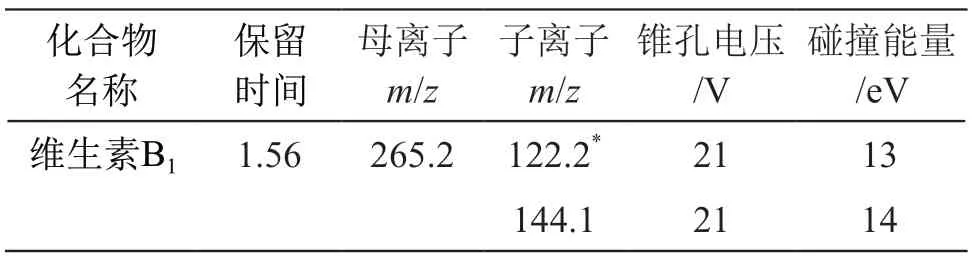
表1 目标化合物的质谱参数
3.3 UPLC-MS/MS色谱条件优化
维生素B1是极性较强的化合物,在一般的C18反相色谱柱上的保留能力差,HPLC中使用的离子对试剂不适合质谱检测系统。本方法选择对极性化合物的保留显著增强的HSS T3色谱柱(100 mm×2.1 mm,1.8 μm),使维生素B1得到了有效的保留和分离,提取离子色谱图见图2。

图2 维生素B1的MRM色谱图
3.4 标准曲线
将标准工作溶液按优化后的色谱条件进样分析,以质量浓度为横坐标,峰响应值为纵坐标绘制标准曲线,见表2。结果表明,两种方法的线性关系均良好。
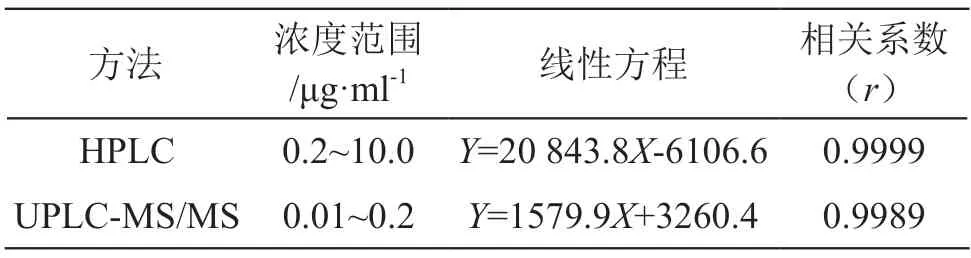
表2 两种方法的线性参数表
3.5 样品检测方法的选择
样品为国际能力验证样品,未指定检验方法,分析现行国标检测方法:GB/T 5009.197-2003直接用甲醇-水-磷酸混合溶液稀释样品后进行检测,无水解和酶解过程,如果样品中含结合态维生素B1,可能提取不完全;GB 5009.84-2016第一法HPLC法样品溶液需衍生后用荧光检测器进行检测,实验过程长且衍生化合物稳定时间短,当有其他荧光物质干扰时可能影响检测准确性;液相色谱-紫外检测器检测样品中维生素B1不需要进行衍生,实验过程简便快捷,质谱检测器在灵敏性、准确度和选择性等方面具有明显优势,能对目标化合物进行准确定性。综上,在国标方法的基础上优化了前处理方法,参照GB 5009.84-2016《食品安全国家标准 食品中维生素B1的测定》[9]中试液提取方法进行样品提取,舍弃后续衍生后用荧光检测器进行检测的方法,采用HPLC-紫外检测和UPLC-MS/MS两种方法进行检测,两种方法的测试结果见表3。由表3可见,两种方法的检测结果无显著差异。
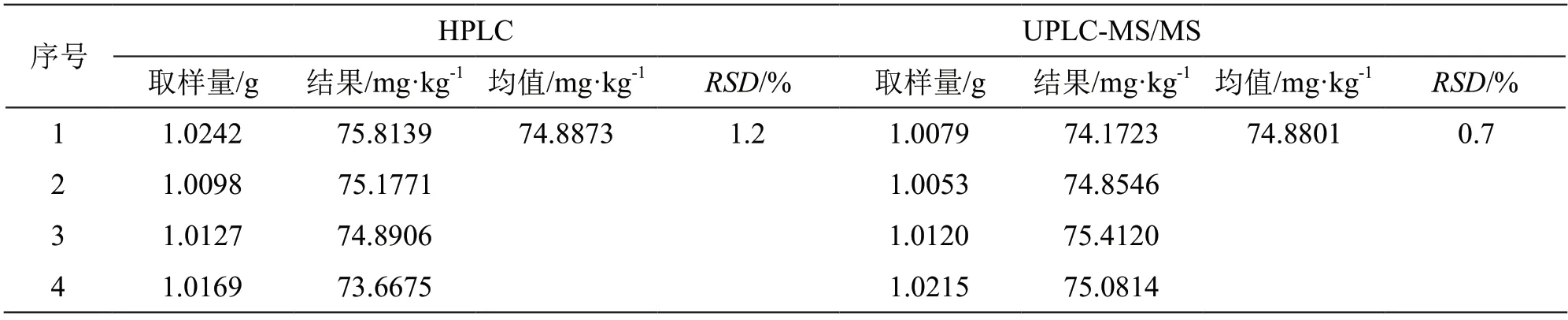
表3 两种方法样品中维生素B1的含量检测结果(n=4)
3.6 加样回收率
为了验证两种方法的准确性,进行了加样回收率的测定,结果见表4。HPLC加标回收率为97.48 %~99.55 %,RSD为0.9 %,UPLC-MS/MS加标回收率为100.43 %~104.33 %,RSD为1.8 %,结果见表4。由表4可见,两种方法加样回收率高,精密度好,表明选择的方法准确、可靠。

表4 两种检测方法加样回收率考察结果(n=4)
3.7 结果上报
按上述结果,最后上报两法的平均值7.49 mg/100 g。本次FAPAS比对,共30个实验室提交维生素B1的数据,30个数据的中位值为7.74 mg/100 g,上报的数据Z值为-0.4,结果为“满意”(“满意”要求∣Z∣≤2),表明本文方法科学、可靠。
4 结论
本研究建立了HPLC和UPLC-MS/MS法同时测定FAPAS液体维生素补充剂能力验证样品中维生素补品中的维生素B1含量,最后上报数值获得“满意”结果。方法对现行国标做了一定创新和改进,经过验证结果准确可靠,具有一定的应用推广价值。
猜你喜欢
中国土壤与肥料(2021年5期)2021-12-02
食品安全导刊(2021年20期)2021-08-30
今日农业(2020年22期)2020-12-14
中国交通信息化(2017年9期)2017-06-06
工业设计(2016年11期)2016-04-16
当代化工研究(2016年5期)2016-03-20
特产研究(2014年4期)2014-04-10
电力需求侧管理(2014年4期)2014-03-20
河南科技(2014年22期)2014-02-27
