
液相色谱-氢化物发生-原子荧光光谱法测定中药样品中的4种砷形态
2024-01-16 03:19刘德晔张琳昀
食品与药品 2023年6期
张 雯,徐 丽,刘德晔,张琳昀*
(1.江苏省疾病预防控制中心,江苏 南京 210009;2.驻马店市食品药品检验所,河南 驻马店 463000)
中药材中的重金属及有害元素的污染一直是国内外普遍关心的问题[1]。随着检测技术的进步,仅测定中药材中的重金属总量已不能满足监测其毒性的要求。同一元素的不同化学形态其毒性存在很大差异[2]。砷是一种广泛存在于自然界中的重金属[3-4],砷对人体的毒性不仅与其总量有关,更与其存在形态有关。砷存在的主要形态可分为有机砷和无机砷两大类,无机砷主要包括砷酸盐[As(V)]和亚砷酸盐[As(III)],有机砷包括一甲基砷(MMA)和二甲基砷(DMA)等,无机砷的毒性比有机砷的毒性强,而在海洋生物中富集较多的有机砷如砷胆碱(AsC)和砷甜菜碱(AsB)等毒性相对较小[5]。研究发现,砷各种形态的毒性大小依次为As(III)>As(V)>MMA>DMA>AsB。无机砷化合物已被国际癌症研究机构确认为I类致癌物[6]。无机砷摄入过量易导致皮肤、神经、消化、生殖、免疫系统等损伤,更加严重的会导致肝癌、皮肤癌等癌症的发生[7]。2020年版《中华人民共和国药典(一部)》规定了 28种中药材及6个中成药中重金属及有害元素限量,《药用植物及制剂 进出口绿色行业标准》规定中药重金属的一般限量标准为砷≤2.0 mg/kg[8-9]。考虑到无机砷的高毒性,中药中砷的限量标准显然不够完善。
常用的砷形态的测定方法主要是食品安全国家标准GB5009.11-2014[10]中的第一法液相色谱-原子荧光法(LC-AFS)[11-12]和第二法高效液相色谱-电感耦合等离子体质谱法(LC-ICP-MS)[13-15],以上两种方法亦可作为中药材中无机砷测定方法。除此之外还有高效液相色谱-电感耦合等离子体串联质谱法(HPLC-ICP-MS/MS)[16-17],但由于仪器昂贵,运行成本较高,难以被普及。本研究在国标第一法LC-AFS方法上进行改进,优化了前处理条件,采用Princen砷形态快速分析柱,选择合适的试剂条件和仪器参数,建立一种能同时测定中药材样品中As(III)、As(V)、DMA和MMA 4种砷形态的方法,对完善中药材中无机砷的限量标准,正确评估土壤环境中砷的污染程度有重要价值,也可为食品和环境监管提供数据支持。
1 仪器与试药
1.1 仪器
DHG-9076A型电热恒温鼓风干燥箱(上海精宏);DEENA3型全自动石墨消解仪(上海仪真);3-18ks型高速离心机(德国Sigma公司);E180H型超声波清洗机(德国Elma公司);Elspe-2型液相色谱仪(广州谱临晟);BAF-4000型原子荧光光谱仪(北京宝德)。
1.2 试药
砷酸根[A s(V)]标准溶液(编号:GBW08667,17.5 μg/g),亚砷酸根[As(III)]标准溶液(编号:GBW08666,75.7 μg/g),MMA标准溶液(编号:GBW08668,25.1 μg/g),DMA标准溶液(编号:GBW08669,52.9 μg/g,中国计量科学研究院);盐酸,硝酸(优级纯,南京化学试剂股份有限公司);磷酸氢二钾(分析纯,南京化学试剂股份有限公司);氢氧化钾,氨水,硝酸铵(分析纯,国药集团);硼氢化钾(分析纯,旭日成化学);高纯氩气(纯度≥99.99 %,南京文达特种气体有限公司);0.45 μm水系滤膜;50 ml聚丙烯离心管。实验用水均为超纯水。本研究所用的中药材均为河南省驻马店市市场监督管理局抽检样品。
2 方法与结果
2.1 LC-AFS条件
2.1.1 LC条件 色谱柱:Princen砷形态快速分析柱(As Spec Fast Analysis Column,4.6 mm×50.0 mm,5.0 μm);保护柱:Princen保护柱(As Spec Guard Column,4.6 mm×50.0 mm,10.0 μm);载气:氩气,压力:0.15 MPa。流动相:流动相A:5 mmol/L 磷酸氢二钾+1 mmol/L硝酸铵,氨水调节pH 10.9;流动相B:25 mmol/L磷酸氢二钾+40 mmol/L硝酸铵,氨水调节pH 9.2; 梯度洗脱程序:0~102 s,100 %流动相A;103~241 s,100 %流动相B;242~360 s,100 %流动相A。流速:1.2 ml/min;进样量:100 μl。
2.1.2 AFS检测条件 载流:5 %盐酸溶液;还原剂:0.5 %氢氧化钾溶液+1.5 %硼氢化钾溶液;负高压:290 V;灯电流:60 mA;辅助电流:60 mA;载气流速:400 ml/min;辅助气流速:800 ml/min。氢气发生器流量:100 ml/min。
2.2 4种不同形态砷含量测定法
2.2.1 标准溶液的配制 准确称取亚砷酸根[As(III)]1.3210 g、砷酸根[As(V)]5.7150 g、MMA 3.9880 g、DMA标准溶液1.8904 g,分别置于10 ml量瓶中,加水定容。所得溶液中As(III)、As(V)、MMA、DMA浓度均为10 μg/ml。分别准确吸取各形态砷标准溶液(10 μg/ml)各1 ml于10 ml量瓶中,加纯水定容,得1.0 μg/ml混合标准溶液(以不同形态的砷计)。
2.2.2 不同形态砷含量测定 称取烘干粉碎后的中药试样0.5 g,置于50 ml消解罐中,加入0.15 mol/L硝酸溶液20 ml,浸泡过夜,经全自动石墨消解仪90 ℃热浸提2.5 h,每0.5 h振摇1 min。提取完毕后,提取液冷却至室温,8000 r/min离心10 min,吸取上层清液,0.45 μm滤膜过滤后上机测定。同时按同一操作方法制备空白溶液。按2.1.1项下色谱条件进样,按2.1.2项下条件检测。
2.3 总砷测定
参考GB 5009.11-2014《食品安全国家标准 食品中总砷及无机砷的测定》中的原子荧光光谱法,称取烘干粉碎后的中药试样1.0 g,置于坩埚中,加入硝酸镁溶液,混匀,再加入1 g氧化镁覆盖样品,于电炉上炭化至无黑烟后,移入550 ℃马弗炉灰化4 h,待样品冷却后加入盐酸溶液中和氧化镁并溶解灰分,转入25 ml量瓶中,加入2 ml硫脲-抗坏血酸溶液后定容至刻度,混匀放置后,按2.1.2项下条件测定总砷含量。
2.4 检测条件的优化
2.4.1 前处理方法的优化 GB 5009.11-2014液相色谱-原子荧光光光谱法[10]测定无机砷中,样品提取方式为90 ℃热浸提2.5 h,每0.5 h振摇1 min,需在干燥箱加热,手动振摇。采用全自动石墨消解仪,可自动设置程序完成升温、每0.5 h振摇1 min以及降温的整个过程,试验结束后自动冷却,自带的特氟龙消解罐能四周加热,保证受热均匀,且无需人为操作。以当归为对象,对比以往文献中使用较多的60 ℃超声加热1 h[16]、90 ℃干燥箱热浸提2.5 h及90 ℃全自动石墨消解仪提取2.5 h 3种提取方式,计算3种方式下提取出的砷的总量,结果见图1。结果表明,60 ℃超声加热1 h提取效率最低,90 ℃干燥箱热浸提及全自动石墨消解仪提取效果相当,由于全自动石墨消解仪操作方便,综合考虑,选择此种消解方式。

图1 不同提取方式下的4种砷形态测定值
2.4.2 色谱柱的优化 试验对比了两种色谱柱,即Hamilton PRP-X100阴离子交换色谱柱(250 mm×4 mm)和Princen砷形态快速分析柱(150 mm×4 mm)对同一浓度的无机砷标准品的分析结果。结果显示两种色谱柱均能有效分离4种砷形态组分,但出峰时间差异较大。两种色谱柱各组分的出峰时间对比见图2。使用Hamilton PRP-X100阴离子交换色谱柱,4种砷形态出峰时间需近1200 s,分析时间较长,而使用Princen砷形态快速分析柱可将分析总时间控制在360 s内,可见Princen砷形态快速分析柱分析速度快,检测效率高,且各组分分离良好。
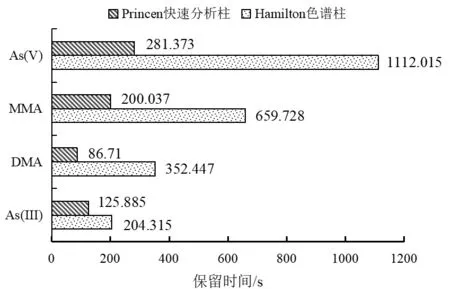
图2 两种色谱柱各组分出峰时间对比
2.4.3 流动相的选择 处理后样品中的砷是以阴离子形态存在的,实验应选择阴离子交换色谱柱进行分离。基于国标方法,选择磷酸盐溶液作为流动相,流动相的pH值是决定分离效果的影响因素之一。由于经过前处理后的样品溶液呈酸性,进入色谱柱会影响样品的保留能力和分离度,为了中和样品的酸性,选择碱性的流动相可达到更好的分离效果,同时可延长色谱柱寿命。结果显示,流动相的碱性较弱不利于峰的分离,若碱性过强(加入氨水调节pH值),则本底值上升。综合考虑,确定流动相A:5 mmol/L磷酸氢二钾+1 mmol/L硝酸铵,pH 10.9;流动相B:25 mmol/L磷酸氢二钾+40 mmol/L硝酸铵,pH 9.2,采用梯度洗脱,保证良好的分离效果。
2.4.4 AFS检测条件的优化
2.4.4.1 载流的浓度 实验过程中,载流与硼氢化钾反应生成氢气,将三价砷还原成气态氢化物,一般选择盐酸溶液。以20 μg/L混合标准溶液为考察对象,分别考察了3 %,4 %,5 %,6 %,7 %盐酸溶液作为载流对待测物峰面积的影响,结果见图3。盐酸浓度为3 %时,4种待测物峰面积明显偏低,随着浓度增加,峰面积逐步上升,后趋于平衡,以6 %,7 %盐酸为载流时的峰面积与以5 %盐酸为载流时基本保持一致,证明5 %盐酸溶液即可满足要求,故实验选择5 %盐酸溶液为载流。

图3 盐酸浓度对4种砷形态峰面积的影响
2.4.4.2 硼氢化钾的浓度 确定用5 %盐酸溶液作为载流后,以20 μg/L混合标准溶液为考察对象,分别考察了氢氧化钾溶液浓度为0.5 %,以1 %,1.5 %,2 %,2.5 %,3 %硼氢化钾溶液作为还原剂对峰面积的影响,结果见图4。由图4可见,随着硼氢化钾浓度的升高,4种砷形态的峰面积明显增大,当浓度达1.5 %后,峰面积趋于平稳,说明1.5 %硼氢化钾溶液即可满足要求,故选择1.5 %硼氢化钾溶液为还原剂。

图4 硼氢化钾溶液浓度对4种砷形态峰面积的影响
2.5 方法学考察
2.5.1 专属性实验 分别吸取2.2.1项下4种砷形态混合标准溶液(1.0 μg/ml)及2.2.2项下阴性样品溶液(空白溶液)、当归样品溶液各100 μl,按2.2项下方法进样测定,色谱图见图5。结果表明,4种砷形态成分均达到基线分离,与其他成分间均无明显干扰,表明检测方法的专属性良好。

图5 专属性实验图谱
2.5.2 线性范围及相关系数 精确吸取一定体积的混合标准溶液,分别用0.15 mol/L硝酸溶液配制浓度为0,1.0,2.0,5.0,10.0,20.0,50.0 μg/L的系列标准溶液,按2.2项下方法检测,记录色谱图。分别以4种砷形态的浓度(μg/L)为横坐标,以其对应的峰面积为纵坐标,绘制标准曲线。方法的线性方程和相关系数见表1。在选定的色谱条件下,标准曲线在1~50 μg/L范围内线性良好,相关系数(r)均>0.999。
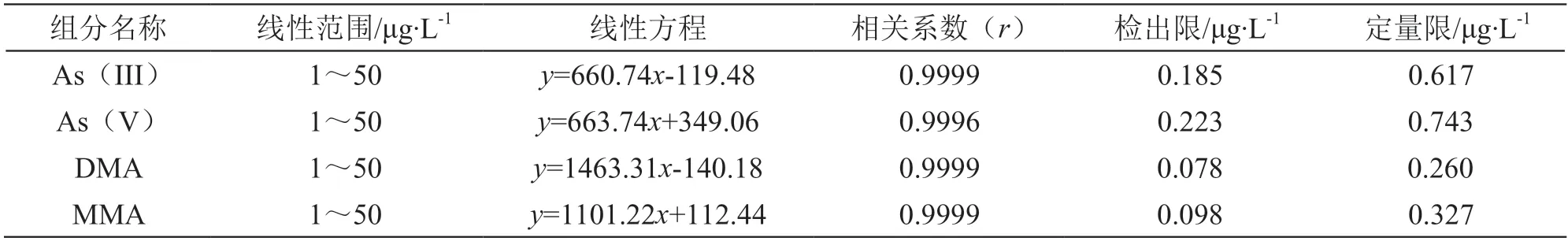
表1 方法的线性范围、线性方程、检出限及定量限
2.5.3 检出限和定量限 采用逐级稀释的方式,以3倍基线噪声时砷形态的浓度为检出限(S/N=3),以10倍基线噪声时砷形态的浓度为定量限,结果见表1。实验中如称取固体样品1.0 g,定容至20 ml时,As(III)、As(V)、DMA和MMA的检出限依次为0.00370,0.00446,0.00156,0.00196 mg/kg,定量限依次为0.0123,0.0149,0.00520,0.00654 mg/kg。完全可满足中药样品中无机砷的分析要求。
2.5.4 精密度和回收率试验 精密称取已测得砷含量的当归样品0.5 g,分别准确加入低、中、高3个浓度水平的4种砷形态混合标准溶液,根据已优化的实验条件对加标样品做回收率试验,加标回收率范围为96.7 %~101.9 %。每个浓度制备6个平行样品,进行精密度试验,计算相对标准偏差(RSD),结果见表2。结果表明方法的准确度和精密度均符合检测要求。
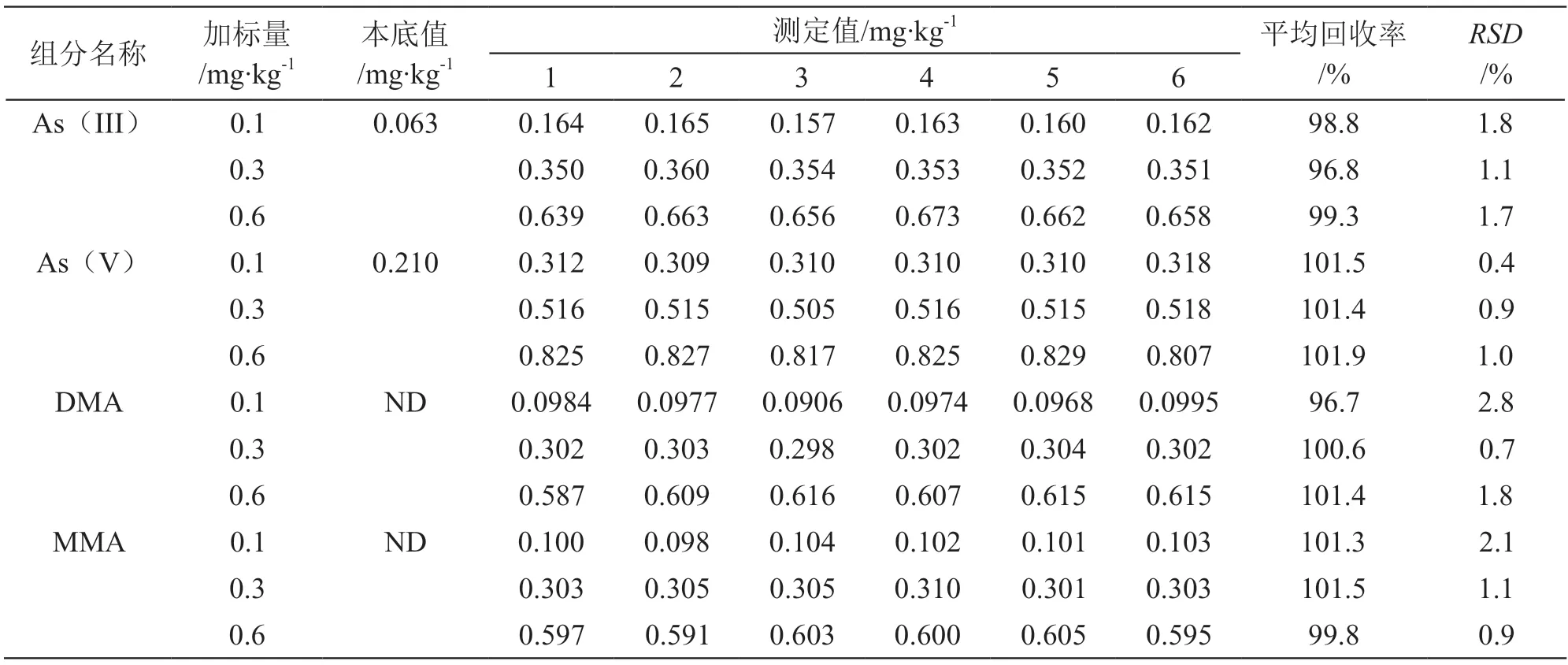
表2 当归样品回收率及RSD值
2.5.5 稳定性实验 取同一批当归样品,加入一定浓度的砷混合标准溶液,按2.2.2项下方法处理,置于4 ℃条件下,分别于0,2,4,6,8,12 h,按2.1.1项下色谱条件进样,按2.1.2项下条件检测。经测算,As(III)、As(V)、DMA、MMA峰面积RSD依次为2.1 %,1.9 %,0.9 %,1.6 %,表明样品溶液在4 ℃条件下放置12 h基本稳定。
2.6 实际样品分析
采用本方法测定市售的7种28批次的中药材中的4种砷形态,同时测定其总砷含量,计算其无机砷含量与总砷含量的百分比。结果见表3。
由表3可见,各批次样品均未检出MMA,仅有黄芪和黄芩中的2批次检出少量DMA,除了甘草1批次未检出As(III)外,其余批次全部检出As(III)和As(V),说明本次检测的中药材中砷形态主要以As(III)和As(V)存在,且As(V)含量均高于As(III),说明中药样品中的无机砷多以As(V)形式存在。2020年版《中华人民共和国药典(一部)》[8]对中药材的重金属限量要求为砷≤2.0 mg/kg,通过检测中药材中的总砷含量,可看出此次检测的中药材中总砷均未超标,无机砷总量占总砷总量的比例为14 %~93 %,表明各样品中不仅存在不同比例的As(III)和As(V),且有未能检出的其他有机砷形态,但由于无机砷的危害远大于有机砷,所以测定样品中的无机砷即可表明中药材的食用风险。
3 结论
本文采用液相色谱-氢化物发生-原子荧光光谱法测定中药材中的4种砷形态。与GB5009.11-2014国标方法相比较,优化了前处理条件,选择了操作更加简便的全自动石墨消解仪,简化了实验操作步骤。同时优选出峰时间更快的Princen砷形态快速分析柱分离样品,使样品在360 s内完全分离,极大地缩短了样品检测时间,节约了试剂和人工成本。本方法检出限低、灵敏度高,各待测组分回收率为96.7 %~101.9 %,RSD均小于3 %,说明方法重复性好,准确度和精密度均能满足方法学要求。综上,本方法在定量分析中药材中As(III)、As(V)、DMA和MMA 4种砷形态中可取得令人满意的结果,值得普及推广。
猜你喜欢
今日农业(2021年12期)2021-11-28
今日农业(2021年7期)2021-11-27
石材(2020年9期)2021-01-07
环保科技(2020年4期)2020-09-03
今日农业(2020年13期)2020-08-24
中国盐业(2018年20期)2019-01-14
分析仪器(2018年5期)2018-10-12
中成药(2016年8期)2016-05-17
山东青年(2016年2期)2016-02-28
华东理工大学学报(自然科学版)(2014年5期)2014-02-27
