
石胆酸通过上调miR-21表达降低核受体PPARα mRNA的稳定性
2024-01-12 06:39:20庞碧滢黄娜娜黄晓霞熊文婷
南方医科大学学报 2023年12期
庞碧滢,黄娜娜,黄晓霞,李 馨,熊文婷,孔 波,姚 焱
广州大学生命科学学院,广东 广州510000
除酒精、病毒、药物等脂肪肝致病因素外,大泡性肝细胞脂肪变性超过5%诊断为非酒精性脂肪性肝病(NAFLD),其疾病谱包括单纯性脂肪肝、非酒精性脂肪性肝炎(NASH)、肝纤维化、肝硬化和肝癌[1,2]。2018年NAFLD全球患病率高达25%[3]。我国NAFLD患病率已赶超欧美发达国家,严重危害国民健康和社会发展[1]。脂肪肝是NAFLD早期阶段,肝脏脂肪分解利用与新生脂肪生成的失调是重要的致病风险因素,因此,促进肝内脂肪酸β-氧化分解,降低肝脏脂肪堆积可有效延缓或阻止NAFLD发展。
过氧化物酶体增殖物激活受体α(PPARα)是肝脏脂质代谢的主要调控蛋白,调控过氧化物酶体β-氧化、线粒体β-氧化、脂肪酸转运与合成等多种代谢途径[4,5]。有研究证实,PPARα激动剂(GW7647)可逆转NAFLD小鼠模型中脂质在肝细胞的积累[6]。此外,NAFLD 患者肝脏PPARα基因表达水平与NASH严重程度和脂肪积累呈负相关,而与NASH 组织学的改善呈正相关[7]。PPARα的调控在NAFLD疾病发展过程中对肝脏有重要的保护作用,以其为靶点的药物Saroglitazar,用于治疗NAFLD/NASH,已进入临床II 期试验,但具体机制尚不明了[8]。
研究表明miR-21-5p[9]、miR-22-3p[10]和miR-34a-5p[11]等miRNAs特异性靶向PPARα,参与PPARα的稳定性调控。其中,miR-21在NASH小鼠模型及患者肝细胞中高量表达,与脂肪变性、炎症和纤维化等过程相关,是NAFLD疾病发展的关键诱导因子[12-15]。miR-21的表达水平受磷酸化的信号转导和转录激活因子3(STAT3)、丝氨酸/苏氨酸蛋白激酶MAPK/ERK和活化蛋白(AP-1)信号通路的调节[12,16]。激活的ERK信号通路介导exportin-5磷酸化影响成熟miRNA水平[17]。现有研究主要集中于miRNA对下游基因的调控,但对于miRNA的上游调节途径不明。
胆汁酸主要由胆固醇衍生而来,在肝脏中形成初级胆汁酸,经肠道细菌转化成次级胆汁酸。在二型糖尿病并发的NAFLD患者和NAFLD疾病的早晚期纤维化进展中,机体大部分胆汁酸水平增加,其中次级胆汁酸石胆酸水平均明显增加[18,19]。肠道菌群失调是NAFLD的重要特征,胆汁酸等肠道菌群代谢物水平和组成的改变常影响NAFLD发病过程[20,21]。有研究表明,肠道菌群可依赖于FXR信号通路诱导肝脏脂肪变性和炎症增加,可通过感应PXR信号通路参与肝脏脂肪酸代谢过程[22,23]。因此,疏水性强的石胆酸可通过肠肝轴激活肝脏FXR和PXR等受体,激活细胞内ERK1/2和JNK1/2等信号通路,以响应肠道菌群带来的变化,影响肝脏代谢功能。
本研究主要从石胆酸与PPARα的mRNA和蛋白表达,转录后调控,以及mRNA稳定性的关系,探究毒性胆汁酸石胆酸诱导MAPK/ERK1/2 信号通路对肝细胞中miR-21水平的调控,及石胆酸通过miR-21影响PPARα mRNA稳定性与肝脏脂质代谢紊乱的可能关系,为NAFLD疾病发展的基础研究和治疗提供新思路。
1 材料和方法
1.1 主要材料与试剂
人肝母细胞瘤HepG2细胞(中科院上海细胞库);ExFect®Transfection Reagent(南京Vazyme);PPARα抗体(Proteintech);phospho-p44/42 MAPK(ERK1/2)(Thr202/Tyr204)抗体(CST);ERK1/2、β-actin、HRP 标记山羊抗兔、HRP 标记山羊抗鼠抗体和Renilla Luciferase(R-Luc)或Firefly Luciferase(F-Luc)报告基因检测试剂盒(上海Beyotime)。
1.2 方法
1.2.1 细胞培养 HepG2 细胞用DMEM 完全培养基(10%胎牛血清,1%青霉素/链霉素)培养(37 ℃,5%CO2),细胞生长至对数期时进行后续实验。
1.2.2 构建phR_hPPARα-3'UTR载体 在NCBI获得人的PPARα(NR1C1,NM_001001928.4)3'UTR 序列,再在TargetScanHuman,miRWalk和DIANA数据库中预测富含miR-21-5p、miR-22-3p和miR-34a-5p等miRNA作用靶点的PPARα 3'UTR序列,按含同一miRNA多个结合位点的序列为一目的片段,得3个短片段,分别命名为3'UTR1(771-1339)、3'UTR2(771-2366)和3'UTR3(4717-5126)。从HepG2细胞总RNA中扩增目的片段,用NotI内切酶将phR载体线性化,按同源重组方法分别构建3个野生型质粒。将3'UTR1中2个miR-21结合序列突变为对应的互补序列,构建突变型质粒:hPPARα-3'UTR1_Mut1、hPPARα-3'UTR1_Mut2 和 hPPARα-3'UTR1_Mut1+Mut2。
1.2.3 单荧光素酶报告基因细胞转染 准备对数期细胞,按ExFect®Transfection Reagent说明书,6孔板每孔转染3 μg质粒,分别将phR_hPPARα-3'UTR载体转染进细胞,8~10 h后,收集细胞。将新鲜的完全培养基进行化合物预处理,分至96孔板,每孔加入50 μL处理后的培养基,每种处理重复3次,再将收集的细胞分至含处理培养基的孔中,每孔分50 μL悬浮细胞,轻轻混匀使细胞均匀铺满培养皿底部。实验处理如下:以50 μmol/L和100 μmol/L 石胆酸为实验处理组,DMSO 为对照组。48 h后,按R-Luc报告基因检测试剂盒说明书,制备蛋白样品,于酶标仪中检测得到每个样品的荧光值。以对照组荧光值/对照组荧光值均值为1,各插入片段的实验组荧光值与对照组荧光值均值之比表示该化合物处理后对该插入片段的调控作用。
1.2.4 RT-qPCR 实验 准备对数期细胞,用DMSO,50 μmol/L和100 μmol/L 石胆酸处理48 h,每种处理重复3 次,按RNA-easyTMIsolation Reagent 试剂盒说明书,提取总RNA,使用随机引物于PCR仪中完成反转录得cDNA。茎环法设计miR-21-5p逆转录引物:5'-GTC GTATCCAGTGCAGGGTCCGAGGTATTCGCACTG GATACGACTCAACAT-3'及qPCR引物,表1中U6引物R 作逆转录引物,配制成混合引物使用。准备总RNA,每10 μL 反转录体系中,250 ng 总RNA、DEPCtreated Water与逆转录引物(each primer 0.5 μmol/L)于PCR 仪70 ℃预变性10 min,立即4 ℃冰浴,再加入其他成分完成反转录(16 ℃30 min;42 ℃30 min;85 ℃5 min;4 ℃hold)。qPCR 扩增均按ChamQ Universal SYBR qPCR Master Mix 说明书,于QuantStudioTM3 System中进行扩增,β-actin为内参基因,U6 snRNA为miRNAs内参基因,目的基因相对表达量以2-ΔΔCt表示。引物序列见表1。
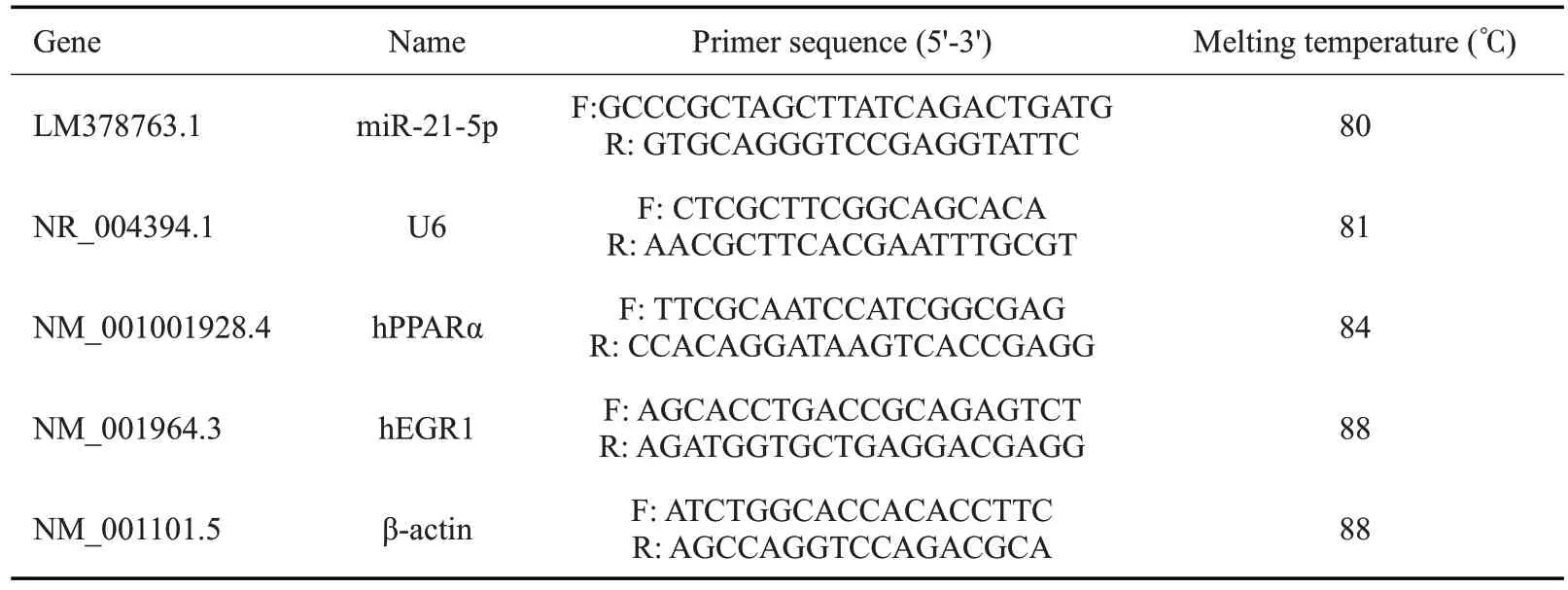
表1 实时荧光定量PCR引物序列Tab.1 Primer sequences for RT-qPCR
1.2.5 miRNAPCR array 在TargetScanHuman、miRDB和DIANA中预测靶向3'UTR1和3'UTR2的miRNAs,取miRDB中Target Score大于50且至少在两个数据库中相互交集的miRNAs为候选miRNAs,以U6 snRNA、SNORD44和SNORD47作内参基因,设置阴性对照,设计miRNA PCR array。以DMSO 为对照,100 μmol/L石胆酸为实验组处理48 h,重复3次,按miRNA cDNA合成试剂盒和miRNAPCR array说明书,各取500 ng总RNA,进行qPCR扩增。以3个内参基因Ct值的均值作标准化,miRNA相对表达量用2-ΔΔCt表示,以log2FC绝对值≥1为差异基因筛选标准。
1.2.6 Western blot 分析 准备蛋白样品,测定蛋白浓度,每孔上样100 μg蛋白进行电泳,转膜(230 mA,90 min),室温封闭2 h(5%脱脂奶粉),洗膜,4 ℃孵育一抗12 h:鼠抗PPARα(1∶1000)和兔抗phospho-p44/42 MAPK(ERK1/2)(Thr202/Tyr204)(1∶500),洗膜,用HRP标记山羊抗鼠或HRP标记山羊抗兔(1∶10000)室温孵育1 h,化学发光显影结束后,膜再生重新封闭,分别4 ℃孵育兔抗ERK1/2(1∶1000)、鼠抗β-actin(1∶1000)过夜,进行后续步骤。数据用Image J处理。
1.2.7 构建pGL4-miPPR-21载体 据已报道的miR-21转录起始点(CATTTTATT)[24],从NCBI 获得VMP1(NM_001329394.2)中hsa-miR-21初始转录本的启动子序列(miPPR-21),经hTFtarget预测确定ERK1/2信号通路下游的主要转录因子—早期生长应答因子(early growth response 1,EGR1)与miPPR-21存在潜在结合作用,TransmiR和JASPAR同时预测到EGR1与miR-21启动子区有潜在靶点(AGACCGCCCCCTCTG,-161~-146)。从基因组DNA中获得目的片段,构建pGL4-miPPR-21(-241~-30)载体,将EGR1与miPPR-21结合位点突变为互补序列,为突变型载体。
1.2.8 双荧光素酶报告基因细胞转染 准备对数期细胞,按转染试剂说明书,6 孔板每孔共转染3 μg 载体(pEnCMV-EGR1 和pGL4-miPPR-21)与20 ng phR 内参质粒,转染8~10 h,收集细胞,将新鲜的完全培养基进行化合物预处理,按每孔50 μL分至96孔板中,每种处理重复3次,再将收集的细胞按每孔50 μL悬浮细胞分至含预处理培养基的孔中,轻轻混匀使细胞均匀铺满培养皿底部。培养24 h后,按F-Luc报告基因检测试剂盒说明书,制备蛋白样品,于酶标仪中检测得到每个样品的萤火虫荧光值(F)和海肾荧光值(R),以对照组F/R与对照组F/R的均值之比为1,实验组每个样品的F/R与对照组F/R的均值之比表示该化合物处理后对插入片段的转录调控,比较该化合物处理后的相对荧光素酶活性变化。
1.2.9 统计学分析 用SPSS和GraphPad Prism 9.0统计并作图。数据表示为均数±标准差,两组间数据采用独立样本t检验分析,P<0.05时认为差异具有统计学意义。
2 结果
2.1 石胆酸下调PPARα mRNA和蛋白水平
100 μmol/L 石胆酸处理HepG细胞48 h后,PPARα mRNA水平显著下调(P<0.01,图1A),同时,在Western blot实验中,PPARα蛋白水平比对照组下调了54%(P<0.01,图1B)。
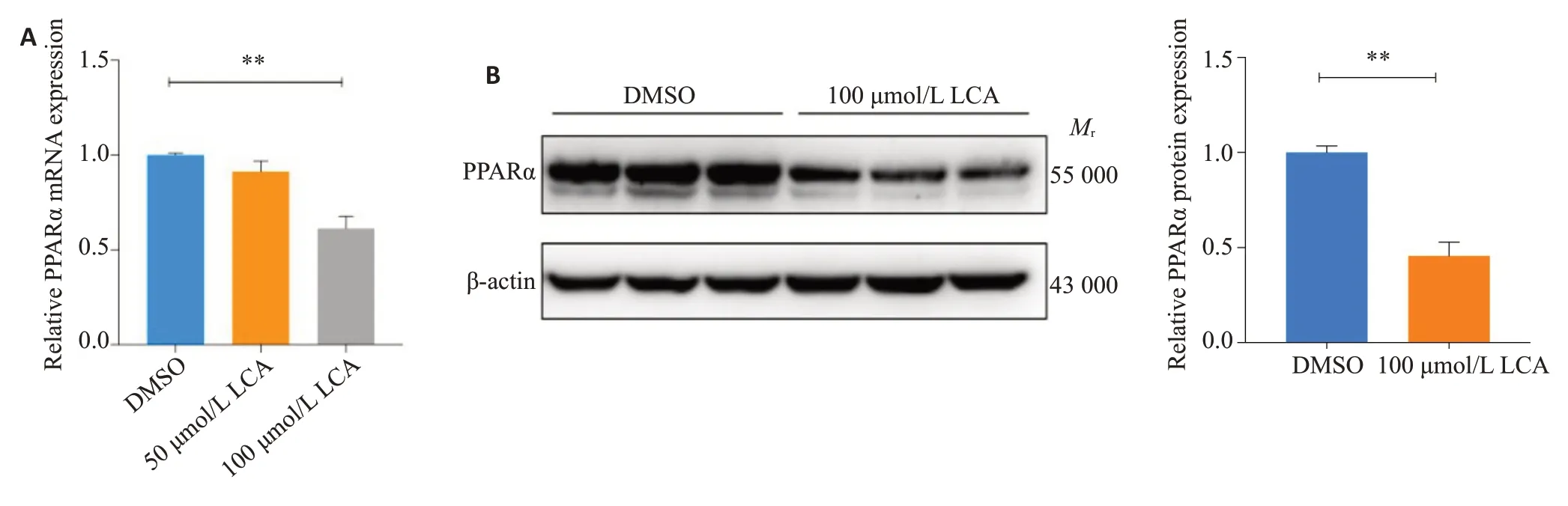
图1 石胆酸下调HepG2细胞中PPARα mRNA和蛋白水平Fig.1 LCA downregulates PPARα at both mRNA and protein levels in HepG2 cells.A:PPARα mRNA levels induced by 50 μmol/Land 100 μmol/LLCAfor 48 h.B:Effect of 100 μmol/LLCAon PPARα protein expression at 48 h.**P<0.01.
2.2 单荧光素酶报告基因体系确证石胆酸参与了PPARα转录后调控
生物信息学预测miRNA靶点丰富的3'UTR区,构建克隆载体(图2A)。与对照组相比,50 μmol/L 石胆酸处理48 h后3'UTR1和3'UTR2的荧光酶活力均显著下降,100 μmol/L 石胆酸处理48 h后,这两个片段的荧光素酶活力均显著下降超过50%(P<0.01),而且降幅均大于3'UTR3(图2B)。
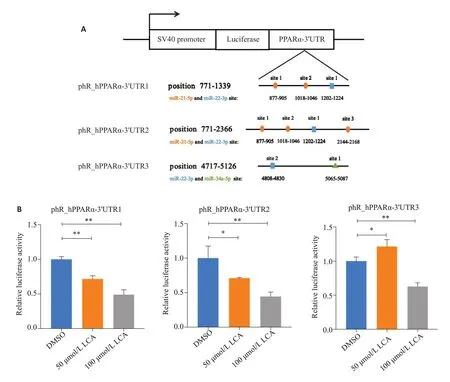
图2 石胆酸对PPARα转录后调控的影响Fig.2 Effect of LCA on post-transcriptional regulation of PPARα.A,B: HepG2 cells transfected with 3 reporter vectors containing binding sequences of miR-21-5p,miR-22-3p and miR-34a-5p and treated with 50 and 100 μmol/L LCAfor 48 h.*P<0.05,**P<0.01.
2.3 石胆酸上调miR-21和miR-22表达
通过生物信息学确定作用于3'UTR1和3'UTR2的候选miRNAs(图3A),100 μmol/L 石胆酸诱导HepG2细胞48 h后,miRNA PCR array检测各候选miRNAs表达水平,以log2FC绝对值≥1为差异基因筛选标准。结果显示,miR-22-3p,miR-21-5p和miR-184的log2FC值分别为1.1,1.0和0.9,则miR-22-3p和miR-21-5p表达上调(图3A)。采用茎环法逆转录,RT-qPCR进一步检测不同浓度石胆酸诱导miR-21-5p表达,其中100μmol/L石胆酸诱导miR-21高水平表达,上调了2.35倍(P<0.05,图3C)。
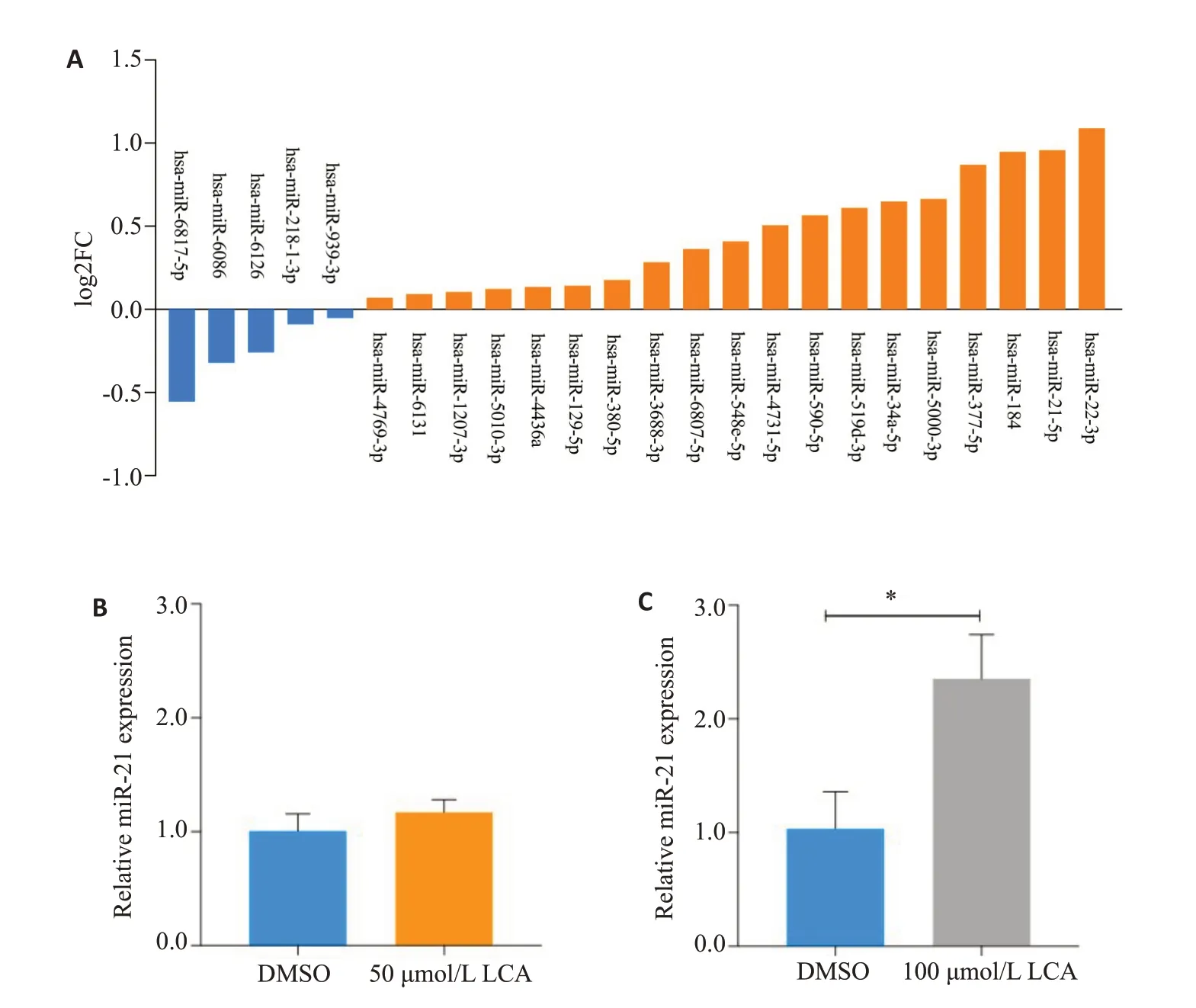
图3 石胆酸上调miR-21和miR-22表达Fig.3 LCA upregulates miR-21 and miR-22 expressions in HepG2 cells.A: Expression changes of candidate miRNAs in miRNA PCR array induced by 100 μmol/L LCA for 48 h.B,C:hsa-miR-21 levels detected by RT-qPCR with Stem-loop reverse transcription induced by 50 μmol/L(B)and 100 μmol/L(C)LCAfor 48 h.*P<0.05.
2.4 石胆酸通过调控miR-21降低PPARα mRNA稳定性
已知miR-21-5p表达上调(图3C),构建3'UTR1中miR-21位点突变载体(图4A)。与对照组相比(图4B),50 μmol/L 石胆酸处理48 h时WT、Mut1和Mut2的荧光素酶活性显著下调,且降幅相当;Mut1+Mut2的荧光素酶活性与对照组相比无显著变化。用100 μmol/L 石胆酸处理48 h(图4C),WT、Mut1和Mut2的荧光素酶活力比对照组显著下调(P<0.01),分别下调51%,37%、39%;Mut1+Mut2 仅下调13%(P<0.05)。显然,两个miR-21位点全部突变后,明显减弱了石胆酸对PPARα的降解。
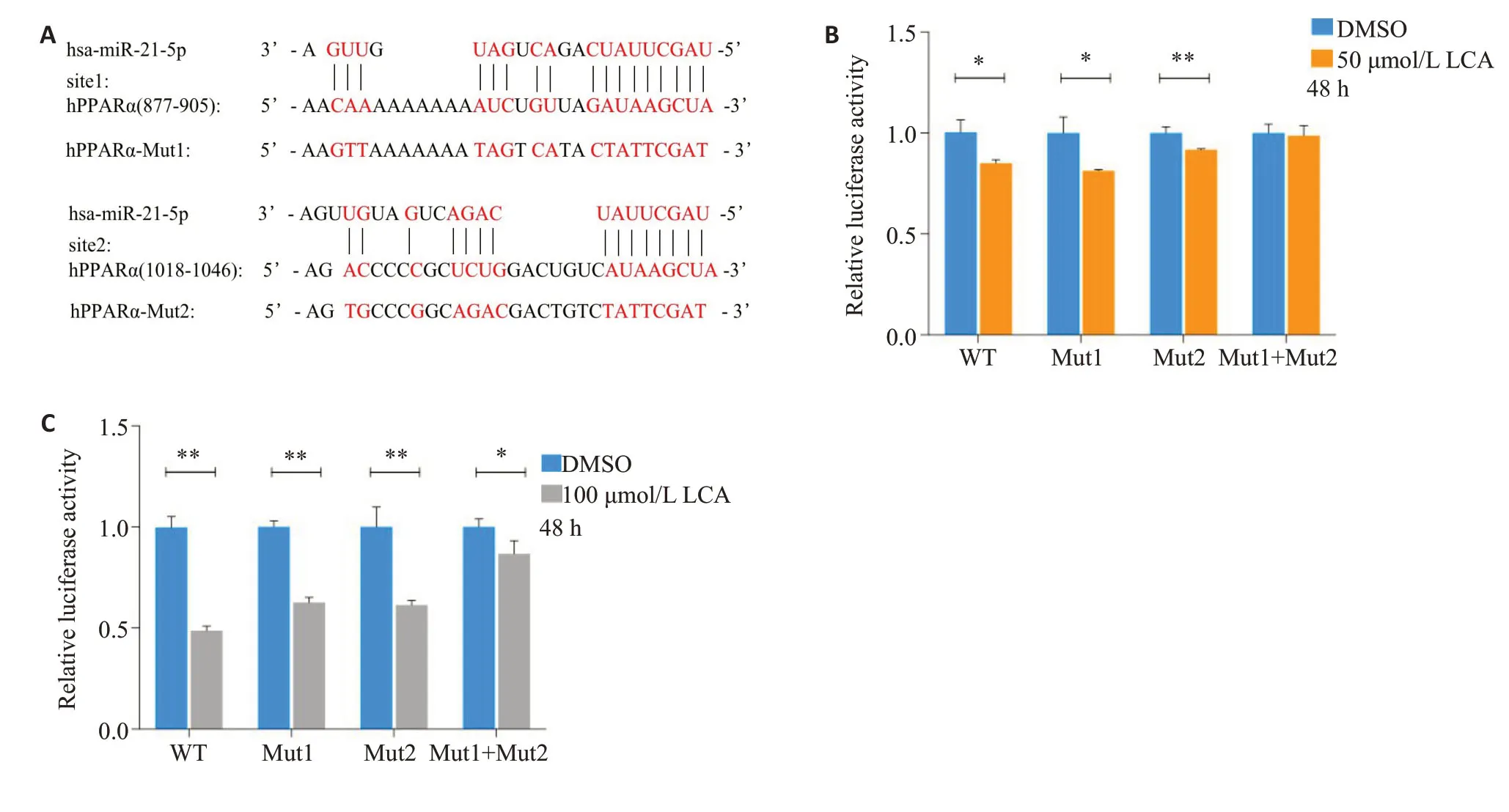
图4 石胆酸通过调控miR-21降低PPARα mRNA稳定性Fig.4 LCA reduces the stability of PPARα mRNA by regulating miR-21.A:Target and mutation sequences of miR-21 in PPARα 3'UTR1.B,C: Changes of luciferase activity after miR-21 sites mutation in cells treated with 50 μmol/L(B) and 100 μmol/L(C) LCA for 48 h.*P<0.05,**P<0.01.
2.5 石胆酸上调转录因子EGR1并影响PPARα蛋白表达水平
Western bolt结果显示:石胆酸在30 min内磷酸化ERK1/2(图5A)。用50 μmol/L 石胆酸处理48 h,EGR1表达上调1.98倍(P<0.05,图5B);100 μmol/L 石胆酸分别处理24 h 和48 h,EGR1 表达上调3.09 倍和5.83 倍(P<0.01,图5C),呈时间剂量正相关。而且瞬时转染过表达EGR1后PPARα蛋白显著下降(P<0.05,图5D),与石胆酸下调PPARα蛋白表达量结果相同(P<0.01,图1B)。

图5 石胆酸调控EGR1对PPARα蛋白表达的影响Fig.5 Effect of EGR1 regulated by LCA on PPARα protein expression.A:Western blots of phosphorylated MAPK/ERK1/2(Thr202/Tyr204)induced by 50 μmol/L and 100 μmol/L LCA for 30 min and 1 h.B,C:EGR1 mRNA expression induced by 50 μmol/L(B)and 100 μmol/L(C)LCA for 48 h.D:Effect of EGR1 overexpression on PPARα protein expression for 48 h.NC:Non-overexpressed EGR1;EGR1 overexpression:Transfection pEnCMV-EGR1.*P<0.05,**P<0.01.
2.6 双荧光素酶报告基因体系确证EGR1与miR-21启动子区的靶向关系
生物信息学预测EGR1 与pri-miR-21 启动子区(miPPR-21)潜在靶点,构建野生型和位点突变型质粒(图6A)。以DMSO对照组酶活性为1,用50 μmol/L 石胆酸处理24 h,不过表达与过表达EGR1时WT的酶活性分别上调,为1.37 和1.58;100 μmol/L 石胆酸处理24 h,不过表达EGR1时WT的荧光素酶活力显著上调1.67,过表达EGR1 时WT 中上调1.90。同时,过表达EGR1时,50 μmol/L 石胆酸处理后Mut的酶活性上升幅度与WT 酶活性升幅相比,呈轻微下降趋势;过表达或不过表达EGR1时,与对照组相比,100 μmol/L石胆酸处理后WT 的酶活性与Mut 的酶活性升幅相当(P<0.01,图6B)。
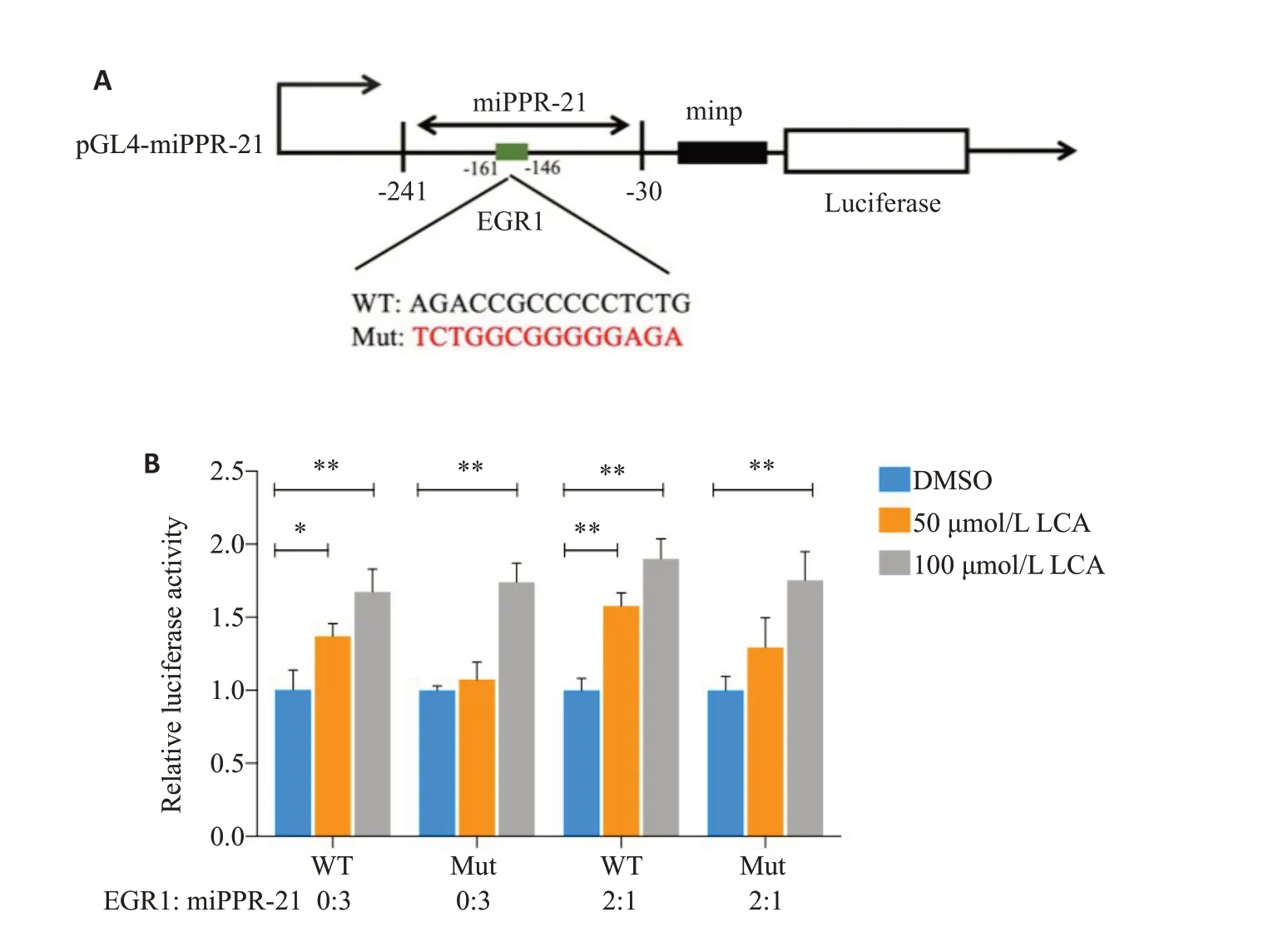
图6 EGR1与miR-21启动子区的靶向关系Fig.6 Targeting relationship between EGR1 and the promoter region of miR-21.A:Vector of pGL4-miPPR-21.B: Effect of EGR1 overexpression on regulation pGL4-miPPR-21 for 24 h.WT:pGL4-miPPR-21;Mut:pGL4-miPPR-21-Mut(-161~-146);EGR1:pEnCMV-EGR1.*P<0.05,**P<0.01.
3 讨论
胆汁酸是营养传感器和代谢调节器,是代谢疾病的重要驱动因素[25]。石胆酸可激活FXR、TGR5、PXR、VDR、EGFR 等受体和JNK1/2、蛋白激酶B(AKT)、ERK1/2等信号通路,参与调控葡萄糖稳态、脂质和脂蛋白代谢、能量消耗、炎症和肝癌的发生[26,27]。临床数据表明NAFLD不同疾病阶段石胆酸水平异常升高,参与NAFLD的疾病进展[18,19]。本研究发现次级胆汁酸石胆酸显著下调肝细胞中PPARα的mRNA和蛋白表达水平,因此有必要了解肝脏中毒性胆汁酸石胆酸对PPARα mRNA稳定性的调控。本文荧光素酶实验证实了石胆酸参与PPARα转录后调控,通过调控富含miRNA 潜在靶点的3'UTR1 和3'UTR2 序列促使PPARα降解。肠道微生物组分析显示高脂饮食小鼠模型中肠道菌群的改变导致石胆酸水平升高,促进NAFLD发展[28]。肝脏中高脂饮食诱导PPARα mRNA表达下调,经降脂药物治疗后表达上调[29]。本文实验结果与其趋势一致,说明肠道代谢物石胆酸可能通过降低PPARα mRNA稳定性影响脂质代谢。
miRNA PCR arrary 筛选发现石胆酸诱导肝细胞miR-21和miR-22差异表达,在本研究中miR-21高水平表达,石胆酸可进一步诱导miR-21表达上调,表明石胆酸对下游基因的调控作用主要通过调节miR-21水平变化来实现。miR-21是肝细胞癌和以脂肪变性为特征的早期肝病中表达丰度高的miRNAs之一,其表达水平的改变与肝脏脂肪变性、脂质过氧化、胰岛素抵抗以及癌症的发生有关[30,31]。miR-21在NAFLD疾病的肥胖、肝炎、肝纤维化及肝硬化阶段中异常高表达,并随NAFLD发展而升高,可促进NAFLD向NASH发展,加速NAFLD进程[32,33]。miR-21表达增加,发生脂质堆积,并直接靶向脂肪酸氧化关键基因PPARα,降低PPARα蛋白表达量[9]。但miR-21其上游调节来源途径不明确。本研究确证了石胆酸对PPARα 3'UTR存在明显的调控作用,并呈剂量相关。生物信息学预测到3'UTR1中miR-21 与PPARα的2 个靶点,靶点突变实验表明miR-21主要通过两个位点起作用,进一步证明石胆酸可通过诱导miR-21作用于PPARα 3'UTR1,在转录后水平降低PPARα mRNA稳定性和蛋白表达,证实了miR-21调控PPARα 3'UTR区的上游石胆酸调节途径。可为解释“NAFLD不同疾病阶段石胆酸水平异常升高”和“高丰度的miR-21促进从肥胖、肝炎到肝纤维化和肝癌的NAFLD疾病发展”的现象提供有力支撑。
MEK/ERK/miR-21在非小细胞癌和癌相关成纤维细胞中表达增加[34],结直肠癌细胞中石胆酸通过ERK1/2信号通路,转录因子AP-1 和STAT3 启动miR-21 转录[35]。本研究中石胆酸可使肝细胞的ERK1/2磷酸化,高剂量石胆酸可快速激活肝细胞中MAPK/ERK1/2信号通路,与石胆酸上调肝细胞miR-21 表达实验结果相呼应,说明肝细胞中ERK1/2 信号通路可能与miR-21 表达相关。研究表明ERK1/2 信号通路下游主要转录因子EGR1在胆汁淤积性肝病患者中的表达增强[36]。本文进一步发现石胆酸诱导HepG2细胞的EGR1 mRNA表达,并呈时间剂量相关,与石胆酸磷酸化ERK1/2结果相同。因此推测转录因子EGR1参与调控miR-21的转录和PPARα表达。过表达EGR1后的PPARα蛋白相对表达量与石胆酸处理后的检测结果一致,均下调PPARα蛋白表达,证实石胆酸诱导EGR1参与PPARα蛋白表达调控的过程。生物信息学分析预测EGR1与miPPR-21有潜在靶点(-161~-146),距转录起始位点较近。双荧光报告基因实验表明EGR1 参与miR-21 转录的过程,在转录后水平对降低PPARα mRNA稳定性有一定的作用,但突变该靶点后,没有减弱过表达EGR1对miPPR-21片段的调控,可见该靶点不是EGR1与miPPR-21的主要结合位点。由过表达EGR1下调PPARα蛋白表达推测,EGR1可能从转录、转录后和翻译水平多方面调控PPARα表达。研究表明,EGR1可通过直接抑制脂肪甘油三酯脂酶的转录,抑制脂肪分解[37]。脱氧胆酸DCA-EGR1-炎症因子轴途径增强CCl4 诱导的肝损伤与PPARα信号相关[38]。可见EGR1与胆汁酸代谢失调、脂质代谢异常和PPARα信号通路密切相关。进一步研究转录因子EGR1影响肝细胞中PPARα mRNA 稳定性和基因表达的具体分子机制,将有助于了解石胆酸下游信号调控PPARα mRNA稳定性的复杂机理。
综上所述,本研究发现石胆酸诱导肝细胞miR-21和miR-22表达上调,并通过诱导miR-21作用于3'UTR参与PPARα转录后调控,促进PPARα降解;石胆酸激活MAPK/ERK1/2信号通路,上调转录因子EGR1并降低PPARα蛋白表达;石胆酸通过MAPK/ERK1/2信号通路及调控miR-21 和miR-22 表达,在转录后水平降低PPARα mRNA稳定性,下调肝细胞PPARα mRNA和蛋白水平,加重肝损伤。本文结果为深入探讨胆汁酸代谢失调在NAFLD疾病发展中的作用机制提供了实验依据。
猜你喜欢
现代临床医学(2022年4期)2022-09-29 07:38:08
理化检验-化学分册(2021年10期)2021-11-29 14:50:42
天津医科大学学报(2021年4期)2021-08-21 02:14:50
中日友好医院学报(2021年1期)2021-04-14 01:58:32
肝博士(2020年5期)2021-01-18 02:50:26
中西医结合肝病杂志(2020年2期)2020-10-27 02:18:42
山东医药(2020年9期)2020-05-20 01:12:16
中国卫生标准管理(2015年16期)2016-01-20 09:26:29
中国当代医药(2015年9期)2015-03-01 02:02:12
中国医药导报(2015年27期)2015-02-28 22:08:01
