
钴酞菁/多孔碳气凝胶复合结构用于高效电催化CO2 还原
2024-01-06 14:26:32龚善和吕晓萌冯玉祥黄春霞朱桂生
煤炭与化工 2023年11期
周 暾,龚善和,吕晓萌,冯玉祥,黄春霞,朱桂生
(1.江苏索普化工股份有限公司,江苏 镇江 212006;2.江苏大学环境与安全工程学院 江苏 镇江 212013;3.江苏大学化学化工学院,江苏 镇江 212013;4.江苏索普聚酯科技有限公司,江苏 镇江 212000)
0 引言
随着全球工业化的进程,过量化石燃料转化成CO2,并排放于空气当中,引起了严重的温室效应,引发了一系列的环境问题。目前,日益严重的环境问题和全球能源紧缺问题已经引起了全世界的重点关注,然而,仅凭植物光合作用难以平衡环境中的碳循环,因此迫切需要寻找一种类“自然界碳循环”的方式,来协助这一问题的解决。通过可再生能源转换(如,风能、光能、潮汐能等)的电能,将CO2电化学还原成化学品和燃料,实现可再生能源与化学能源的耦合,从而减少温室气体排放,是实现碳中和的新途径。其中,CO 作为简单的两电子转移的产物,已被证明是一种具有实现工业化潜力的产物。因为其本身可直接利用,或再加工生产,以及电化学过程中令人满意的生产率和低电能成本。然而,要实现CO2RR 工业化,仍然受到具有稳定C=O 键的CO2分子的高活化能垒(806 kJ·mol-1),以及CO2分子在水性电解质中的低溶解度的影响。此外,竞争性析氢反应(HER)也对提高还原产物的法拉第效率(FE)提出了巨大挑战。因此,开发高活性、高选择性和高稳定性的电催化CO2还原催化剂仍然是近期研究的热点问题。
1 存在的问题
过渡金属配合物(主要包括大环配体(卟啉、酞菁、环胺等)),多吡啶配体(联吡啶、三联吡啶等),钳形配体(膦等)和卡宾配体等大环金属分子)在均相电催化CO2还原转化为CO 领域中的应用逐渐引起了广泛的关注。过渡金属的d 轨道具备多价氧化还原能力,可满足涉及多个质子耦合电子转移步骤的电催化CO2还原的要求,又可充当多电子反应金属还原中心。但传统的均相催化剂,存在:①溶解性差;②原子利用率低;③电子传输缓慢;④回收利用困难;⑤形成团聚而失活等问题。借助导电材料为基底,均相催化剂为催化单元,构建的多相分子催化剂,能有效解决上述存在的问题。例如,以CoPc 为催化单元,构建的Co 基多相分子催化剂,在电催化CO2还原中,表现出优异的性能。
但目前优化Co 基多相分子催化剂的工作主要集中于酞菁环外功能化修饰对位点电催化性能影响的研究,忽略了基底的孔隙结构对位点活性的影响。然而,分级多孔的结构对促进位点周围CO2传质,提升位点活性,是十分有效且重要的。因此,优化基底孔隙结构,以实现对催化单元微环境的优化,促进位点周围CO2的传质和产物的扩散,对构建高活性、稳定性的催化剂是有必要的。
碳基材料因其丰富的微观结构和形貌、高固有电导率、优异的稳定性、重量轻和低成本而引人注目,已被广泛用作储能材料、催化剂、催化载体、化学吸附剂、热绝缘体和隔音材料。其中,碳气凝胶(CA)因其特殊性质而受到更多关注,包括高比表面积、低密度、丰富的孔隙、可控的结构、纹理、良好的导电性和化学性质。其三维(3D)分级多孔网络骨架有利于客体离子/分子移动到内部结构中,加速催化位点和反应相关物质之间的接触,以促进反应。
借助碳气凝胶特殊的多级孔特性,可优化催化单元周围CO2传质,促进CO2与位点接触;同时,碳气凝良好的导电特性,将优化催化剂导电能力,促进电子界面传导,加快电催化反应,提升基于CoPc 为催化单元的催化剂性能。通过水热合成反应的高温、高压环境,合成了氮掺杂的“类珊瑚状”多孔碳气凝胶。通过非共价键固定的方法,将CoPc 稳定于气凝胶骨架的表面,以实现活性位点和导电碳骨架稳定结合。N-CA 的特殊的多级孔结构促进了Co 位点的暴露,同时,加快位点周围CO2传质。因此,CoPc@N-CA 展现出的优异电催化CO2还原性能,显著高于基于商业CB 而设计的CoPc@CB 催化剂。CoPc@N-CA 阴极催化剂,在较宽的电势窗口(-0.5~-1.0 V vs.RHE)下,对CO的选择性>80%,最高为99.05%,在-0.85 V vs.RHE 电位下,对CO 的部分电流密度达到14.40 mA·cm-2。
2 实验部分
2.1 实验药品
乙醇(CH3CH2OH,≥99.7%)、苯酚(C6H6O,≥99.5%)、甲醛(HCHO,37%)、三聚氰胺(C3H6N6,99%)、乙酸(CH3COOH,99.7%)、N,N-二甲基甲酰胺(DMF,≥99.5%)、碳酸氢钾(KHCO3,≥99.9%)、商品炭黑(CB)均购自国药化学试剂有限公司。钴酞菁(CoPc,≥96%),高纯氮气(N2,99.99%)、高纯二氧化碳气体(CO2,99.999%)和高纯氩气(Ar,99.999%)购自江苏索普气体有限公司(中国,江苏)。
2.2 材料制备
2.2.1 合成氮掺杂的多孔碳气凝胶(N-CA)
将1.882 g C6H6O 和3 mL HCHO 加入到30 mL含有0.6 mL CH3COOH 的去离子水中,然后在70℃下连续搅拌1 h。将含有0.4 mL CH3COOH、0.3 g C3H6N6和537 μL HCHO 的20 mL 去离子水加入到上述溶液中(固体前驱体预溶解后再加入),然后连续搅拌30 min。
将混合溶液放入100 mL 聚四氟乙烯高压釜中,在160 ℃加热16 h,得到水凝胶。将水凝胶浸入丙酮中24 h 以除去不稳定组分,然后,所得样品通过冷冻干燥处理获得干凝胶。所得干凝胶在N2气氛下,800 ℃碳化3 h(升温速率为2 ℃·min-1),然后自然冷却至室温得到N-CA。
2.2.2 合成酞菁钴/氮掺杂的多孔碳气凝胶(CoPc@N-CA)将50 mg N-CA 加入50 mL DMF 溶液中,连续超声30 min 得到N-CA-DMF 悬浊液,标记为A 溶液。将5 mg CoPc 加入50 mL DMF 溶液中,连续超声30 min 得到CoPc-DMF 悬浊液,标记为B 溶液。在剧烈搅拌下,将B 溶液将入到A 溶液中,并持续搅拌24 h。真空抽滤上述悬浊液,再通过DMF清洗几次后置于60 ℃烘箱,烘干,得到CoPc@N-CA 样品。
2.2.3 合成酞菁钴/碳黑(CoPc@CB)
CoPc@CB 合成方法与CoPc@N-CA 相似,除了基底N-CA 被CB 替换。
2.3 电极制备
将6mgCoPc@N-CA 或CoPc@CB,3mLCH3CH2OH 和30 μL Nafion(5 wt%)溶液混合,并超声处理30 min,以获得催化剂油墨。将催化剂油墨溶液滴涂到碳纸(双面涂覆,Toray,TGP-H-60,20%PTFE)的表面上,涂覆面积为1 cm,并干燥以获得工作电极(负载量为1 mg·cm-2)。
2.4 电化学测试
采用三电极系统于H-cell 中测试材料的性能,电信号收集采用电化学工作站(上海,CHI1140c)。置于阴阳极的电极液为0.5 M KHCO3电解质(pH=7.3),H-cell 通过Nafion-117 膜分离,以防止产物的交汇。选择银/ 氯化银电极(3.5 M KCl)作为参比电极,铂(Pt)电极用作对电极。根据反应的几何面积(1 cm2)计算电流密度。所有测得的电位通过以下公式(能斯特方程,25 ℃)转换为可逆氢电极(vs.RHE)。
式中:nx为产生一个产物分子所需的电子数(H,CO=2);F 为法拉第常数(96 485 C mol-1);为CO2鼓泡的流速;υ 为由GC 电流信号的峰面积计算出的气体产物的体积比,为298.15 K;Itotal:稳态测试电流(C/s)。
实验中施加不同的电压,进行恒电压测试。通过配备有热导检测器(TCD)和火焰离子化检测器(FID)的在线气相色谱(Online-GC)分析气体产物,使用DMSO 作为内标,通过1H NMR 核磁检测液体产物。CO2气体的流速由质量流量控制器控制在20 sccm。H 型电解池的阴极区加入转子,搅拌速度为400 r/min,以减少传质阻力并加速电极表面上产生的气泡的溢出。线性扫描伏安法(LSV)测试在扫描速率为5 m·V s-1下进行。电化学活性表面积(ECSA)和电化学阻抗谱(EIS)实验在玻碳电极上进行,玻碳电极的反应面积为0.197 cm2,催化剂涂覆量为0.15 mg·cm-2。在电压区间为0~0.3 V vs.RHE 下,进行ECSA 的测试实验。EIS 实验在0.5~200 000 Hz 的测试频率下进行,测试电位设定为-0.60 V vs.RHE。
2.5 材料表征
用配备有单色Cu Kα 辐射(λ=1.541 78A)的X 射线衍射仪(XRD,德国布鲁克AXS 公司)收集样品的粉末XRD 谱图。拉曼(Raman)光谱由Thermo Electron Corporation DXR 拉曼光谱仪(USA)使用500 nm 激光源获得。X 射线光电子能谱(XPS)光谱由ESCA PHI500 光谱仪检测。场发射扫描电子显微镜(FESEM)图像是用配备有能量色散X 射线光谱(EDS)的场发射扫描电子显微分析仪(日立,Regulus-8100,日本)进行测试的。透射电子显微镜(TEM,日本,JEOL-2100F)在100 kV 下操作。400 MHz 核磁共振1H 谱(BRUKER AVANCE 400)来确定液体产物成分。
3 结果与讨论
3.1 材料形貌表征
以三聚氰胺(M)、苯酚(P)和甲醛(F)为原料合成干凝胶。首先,在酸性条件下,P 和F 通过水热法发生缩聚反应形成PF 聚合物。另一个缩聚反应发生在M 和F 之间,以形成MF 聚合物,MF 和PF 的羟甲基之间的进一步缩聚反应形成亚甲基醚桥连聚合物,后者可以共缩合以形成由支化聚合物种类组成的小球状单元。进一步的水热条件下,交错分子的端基连续生长以形成湿凝胶的三维(3D)聚合物网络结构。通过冷冻干燥处理湿凝胶以获得干凝胶,干凝胶进一步碳化以形成N-CA。通过N-CA 上富电子N 位点作为轴向配体或π-π相互作用,将CoPc 锚定在N-CA 表面,形成CoPc@N-CA 样品。CoPc 通过π-π 相互作用稳定在CB 表面,形成对比样CoPc@CB 材料。
CoPc@N-CA 材料合成机理如图1 所示。

图1 CoPc@N-CA 的合成机理图Fig.1 Synthesis mechanism diagram of CoPc@N-CA
以三聚氰胺(M)、苯酚(P)和甲醛(F)为原料合成干凝胶。首先,在酸性条件下,P 和F 通过水热法发生缩聚反应形成PF 聚合物。另一个缩聚反应发生在M 和F 之间,以形成MF 聚合物,MF 和PF 的羟甲基之间的进一步缩聚反应形成亚甲基醚桥连聚合物,后者可以共缩合以形成由支化聚合物种类组成的小球状单元。进一步的水热条件下,交错分子的端基连续生长以形成湿凝胶的三维(3D)聚合物网络结构。通过冷冻干燥处理湿凝胶以获得干凝胶,干凝胶进一步碳化以形成N-CA。通过N-CA 上富电子N 位点作为轴向配体或π-π相互作用,将CoPc 锚定在N-CA 表面,形成CoPc@N-CA 样品。CoPc 通过π-π 相互作用稳定在CB 表面,形成对比样CoPc@CB 材料。
CoPc@N-CA 的电子显微镜图如图2 所示。

图2 CoPc@N-CA 的电子显微镜图Fig.2 Electron microscope images of CoPc@N-CA
在图2a 中,CoPc@N-CA 的场发射扫描电子显微镜(FESEM)图像显示了具有富孔结构的3D 交联“类珊瑚状”纳米气凝胶网络形态,其保留了N-CA 的原始形态。其中纳米交联单元的尺寸相互连接形成气凝胶网络结构,以及大孔互联的结构。这种特殊的结构将有助于CO2或CO2RR 相关物种在CO 位点周围的大量运输,以提高CO2RR 的活性。在CoPc@N-CA 中没有发现CoPc 晶体残留物,这意味着CoPc 均匀地锚定在N-CA 的表面上。CoPc@N-CA 的透射电子显微镜(TEM)图像清楚地揭示了多孔无序纳米网络结构(图2b)。对应放大的TEM(图像(图2c)未发现聚CoPc 的存在,说明CoPc 均匀分散在N-CA 的表面。
CoPc@CB 的电子显微镜图如图3 所示。

图3 CoPc@CB 的电子显微镜图Fig.3 Electron microscope images of CoPc@CB
使用商业CB 作为碳载体合成的CoPc@CB,通过SEM 和TEM 观察CoPc@CB 的形貌(图3a-b),其仅显示与CB 相似的尺寸不均匀的大块状结构,与CoPc@N-CA 的“类珊瑚状”形成鲜明的对比。同时,CoPc@CB 放大的TEM 图像同样未发现聚CoPc 晶体的存在(图3c),说明CoPc 均匀负载于CB 的表面,与CoPc@N-CA 中观察的结构类似。
CoPc@N-CA 的形貌测试图如图4 所示。

图4 CoPc@N-CA 的形貌测试图Fig.4 Morphology test images of CoPc@N-CA
CoPc@N-CA 的SEM 图像(图4a)和对应的能量色散X 射线光谱(EDS)元素分析图像揭示C(图4b)、N(图4c)和Co(图4d)元素均匀分布在3D 多孔纳米网络上,与上述分析结果一致,说明Co 位点均匀分散于CoPc@N-CA 中。
3.2 结构表征
CoPc@CB 和CoPc@N-CA 的表征测试图如图5所示。
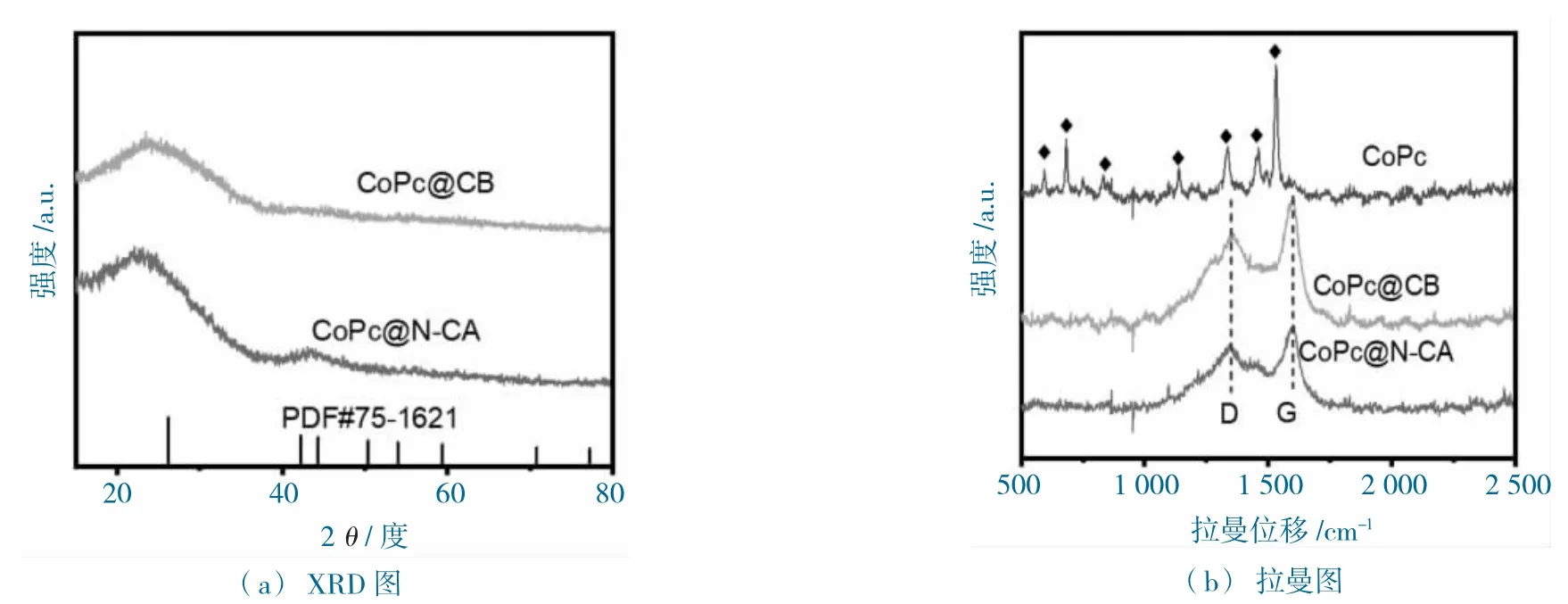
图5 CoPc@CB 和CoPc@N-CA 的表征测试图Fig.5 Characterization test diagram of CoPc@N-CA and CoPc@CB
由CoPc@N-CA 和CoPc@CB 的粉末X 射线衍射(XRD)图分析发现,在23.4 和44.4 处出现2个宽峰,分别属于石墨碳的(002)和(100)无定型晶面(PDF#75-1621,图5a),未发现CoPc 的特征晶体峰,说明CoPc 均匀分散于基底中,这与电镜表征的结果一致。进一步的材料Raman 光谱测试发现,商业的CoPc 在500~1 800 cm-1,显示一系列特征谱峰(图5b),而CoPc@N-CA 和CoPc@CB仅显示G 带(1 585 cm-1)和D 带(1 351 cm-1)2 个肩峰,对应于无缺陷的sp2 碳结构和具有边缘平面缺陷的石墨碳,未发现CoPc 的特性谱峰。
CoPc@CB 和CoPc@N-CA 的XPS 测试图如图6所示。
样品中元素信息和价态通过X 射线光电子能谱(XPS)来分析(图6a)。元素C、N、Co 存在于 CoPc@N-CA 和 CoPc@CB 中,其 中,CoPc@N-CA 中Co 的含量为0.3 at%,而CoPc@CB中Co 的含量为0.29 at%(图6b)。
样品中C 的主峰被矫正为284.8 eV(图6c-d)。CoPc@N-CA 高分辨率XPS N 1s 分析发现,Co-N(399.69 eV)物种的出现,说明CoPc 的存在(图6e)。
同时,CoPc@CB 的高分辨率XPS N 1s 分析中,也同样发现Co-N(400.61 eV)单元的存在(图6f),证明CoPc 稳定于CB 中。相比于商业的CoPc,CoPc@CB 和CoPc@N-CA 中的Co-N 的结合能向高的能级偏移,说明酞菁环与基底之间发生了强电子相互作用,这将有助于稳定CoPc 于基底的表面,优化位点活性。
进一步分析了Co 元素在两个催化剂中的价态,CoPc@N-CA 中Co 2p3/2的位置为780.83 eV(图6g),CoPc@CB 中Co 2p3/2的位置为781.12 eV(图6h),Co 结合能的位置接近Co2+2p3/2(780.84 eV),说明样品中的Co 的价态都为+2 价。
同时,XPS 拟合发现CoPc@N-CA 和CoPc@CB拥有一样的 Co 2p3/2半峰宽,说明 Co 在CoPc@N-CA 和CoPc@CB 中的配位环境是一样的。
3.3 电化学性能测试
CoPc@N-CA 和CoPc@CB 在CO2饱和的0.5 M KHCO3溶液中的电化学性能图如图7 所示。
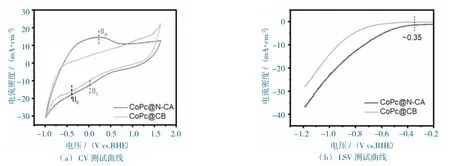
图7 CoPc@N-CA 和CoPc@CB 在CO2 饱和的0.5 M KHCO3 溶液中的电化学性能图Fig.7 The electrochemical performance of CoPc@N-CA and CoPc@CB at CO2-filled 0.5 M KHCO3 electrolyte.
CoPc@N-CA 和CoPc@CB 的CO2RR 的电化学活性测试,在H-cell 中进行(详见实验部分),所有提到的电势都转化为vs.RHE(无iR 补偿),测试电流转化成电流密度。
循环伏安法测试曲线(图7a)所示,CoPc@N-CA 和CoPc@CB 都显示出对CO2的催化活性,这与线性扫描伏安法测试的结果相一致(图7b)。
对比CoPc@CB 的性能,CoPc@N-CA 显示出明显增强的活性(图7b),在相同的电位下,CoPc@N-CA 表现出更大的电流密度,这可能与其多孔、易传质的结构相关。
CoPc@N-CA 和CoPc@CB 的电化学性能图如图8 所示。
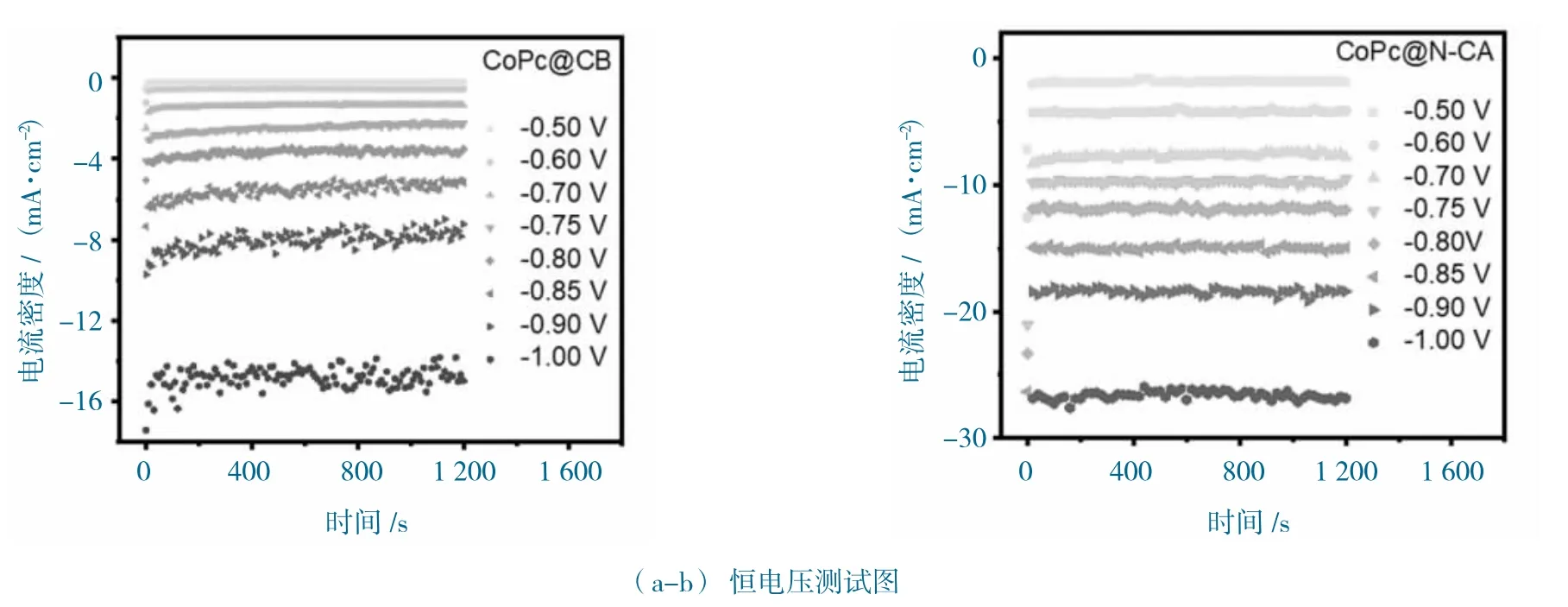
图8 CoPc@N-CA 和CoPc@CB 的电化学性能图Fig.8 The electrochemical performance of CoPc@N-CA and CoPc@CB
进一步恒电位测试,CoPc@CB 和CoPc@N-CA的测试曲线如图8a-b 所示。CoPc@N-CA 在相同的测试条件下,表现出明显增强的活性和稳定性。在-0.9 V vs.RHE 下,CoPc@N-CA 显示-18.49 mA·cm-2的总电流,而CoPc@CB 仅显示-7.68 mA·cm-2的总电流。在线气相分析只发现了H2和CO 产物,1H 核磁分析未发现液相产物。不同电位下对应CO的法拉第效率(FECO),如图8c 所示。CoPc@N-CA和CoPc@CB 的FECO 展现出“类火山”趋势,获得最大值为99.05%(-0.7 V vs.RHE),而CoPc@CB 获得最大CoPc@CB 为89.41%(-0.75 V vs.RHE),这说明N-CA 特殊的结构,可以增强位点对还原产物的选择性。进一步计算CO 的部分电流密度(jCO),CoPc@N-CA 显示出CoPc@CB 更高的jCO,如图8d 所示。在-0.85 V vs.RHE 电位下,CoPc@N-CA 的jCO 达到14.40 mA·cm-2,是CoPc@CB 的4 倍(3.66 mA·cm-2),这说明CoPc@N-CA 特殊的多孔、3D 无序网络结构,有利于促进对CO 产物的活性。CoPc@N-CA 和CoPc@CB 的电化学性能图如图9 所示。
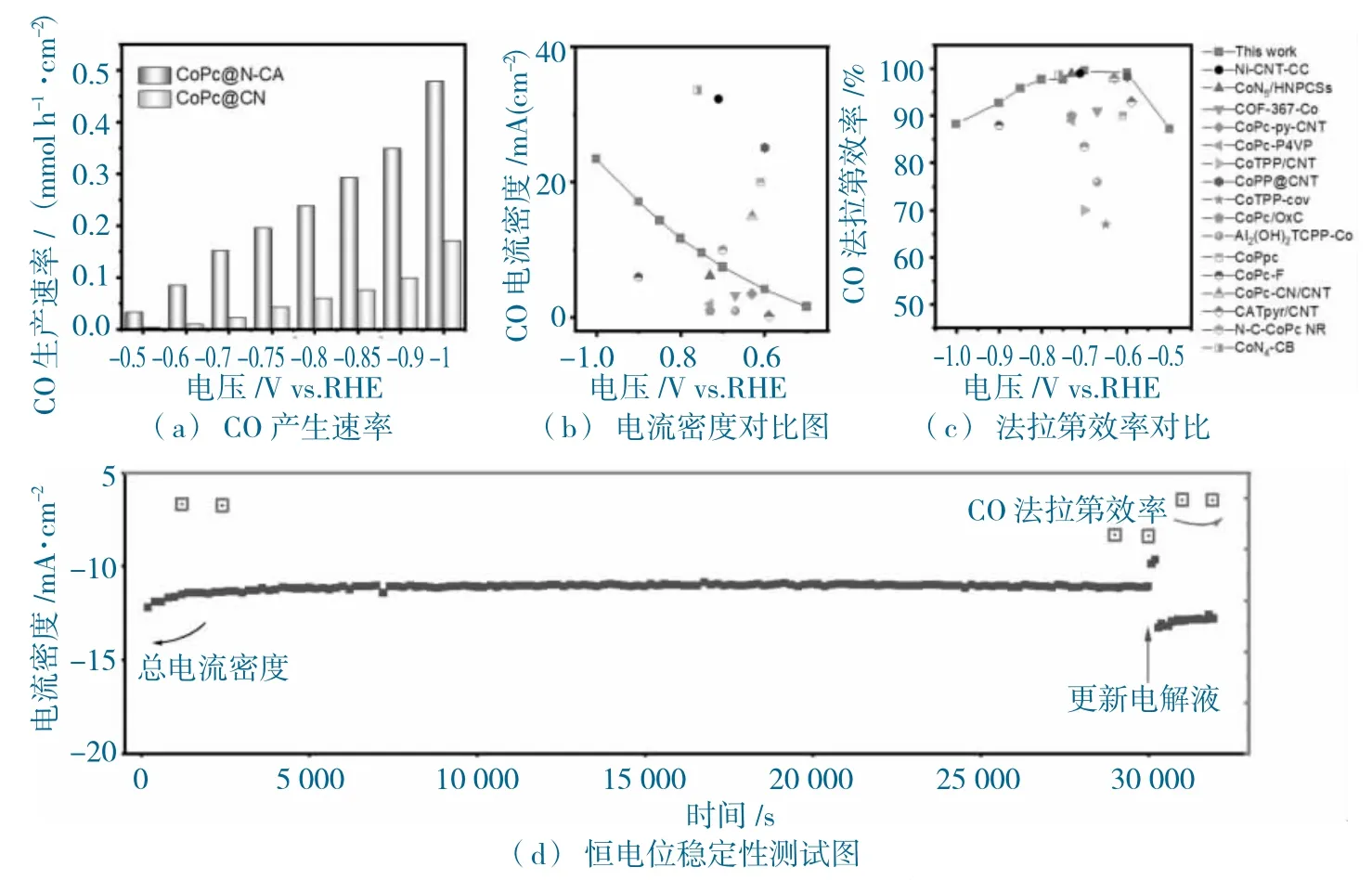
图9 CoPc@N-CA 和CoPc@CB 的电化学性能图Fig.9 The electrochemical performance of CoPc@N-CA and CoPc@CB
在-1.00 V vs.RHE 电位下,在CoPc@N-CA 上产CO 的速率为0.48 mmol h-1cm-2(图9a),而CoPc@CB 仅有0.17 mmol h-1·cm-2。CoPc@N-CA 的jCO(图9b)与FECO(图9c),与已报道的Co 基多相分子催化剂的性能相比,明显高于大多数催化剂的电化学性能。进一步恒电位测试发现(-0.8 V vs.RHE,图9d),总电流稳定在大约-11 mA·cm-2,30 000 s 的测试条件下;,其CO 选择性维持在85%以上,并且通过更新电解池,CoPc@N-CA 的电催化活性可以恢复,如图9d 所示,突出CoPc@N-CA催化剂的高选择性,高稳定性。
3.4 内在活性分析
样品的活性表面积分析图如图10 所示。
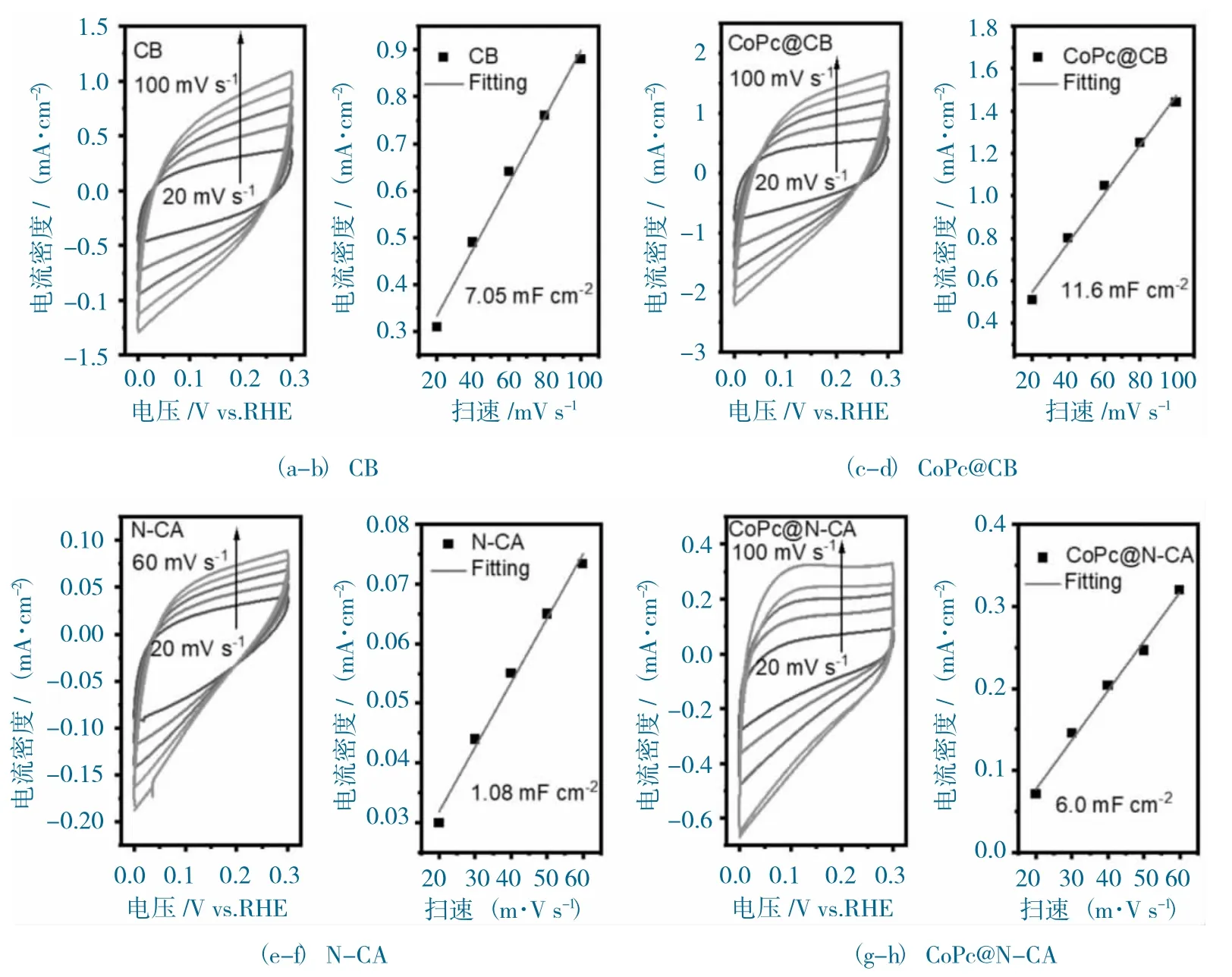
图10 样品的活性表面积分析图Fig.10 The ECSA analysis of samples
测试材料的双层电容(Cdl)值与电化学活性面积(ECSA)成正比,因此通过比较测试催化剂的Cdl 值,可以评估催化剂的活性面积。CoPc@N-CA 和CoPc@CB 的ECSA 测试在非CO2法拉第区间进行。为了探究催化剂真实的ECSA 值,首先测试了基底CB 和N-CA 的Cdl 值,分别为7.05 mF·cm-2(图10a-b)和1.08 mF·cm-2(图10e-f)。CoPc@N-CA 和CoPc@CB 的Cdl 值分别为11.6 mF·cm-2(图10c-d)和6.0 mF·cm-2(图10g-h)。扣除背景的影响,CoPc@N-CA 的Cdl 值应该为4.92 mF·cm-2,而CoPc@CB 的Cdl 值为4.55 mF·cm-2,说明CoPc@N-CA 表面暴露出更多的Co活性位点,这与N-CA 本身3D 多孔“类珊瑚状”的结构相关。
CoPc@N-CA 和CoPc@CB 的阻抗和Tafel 斜率图如图11 所示。
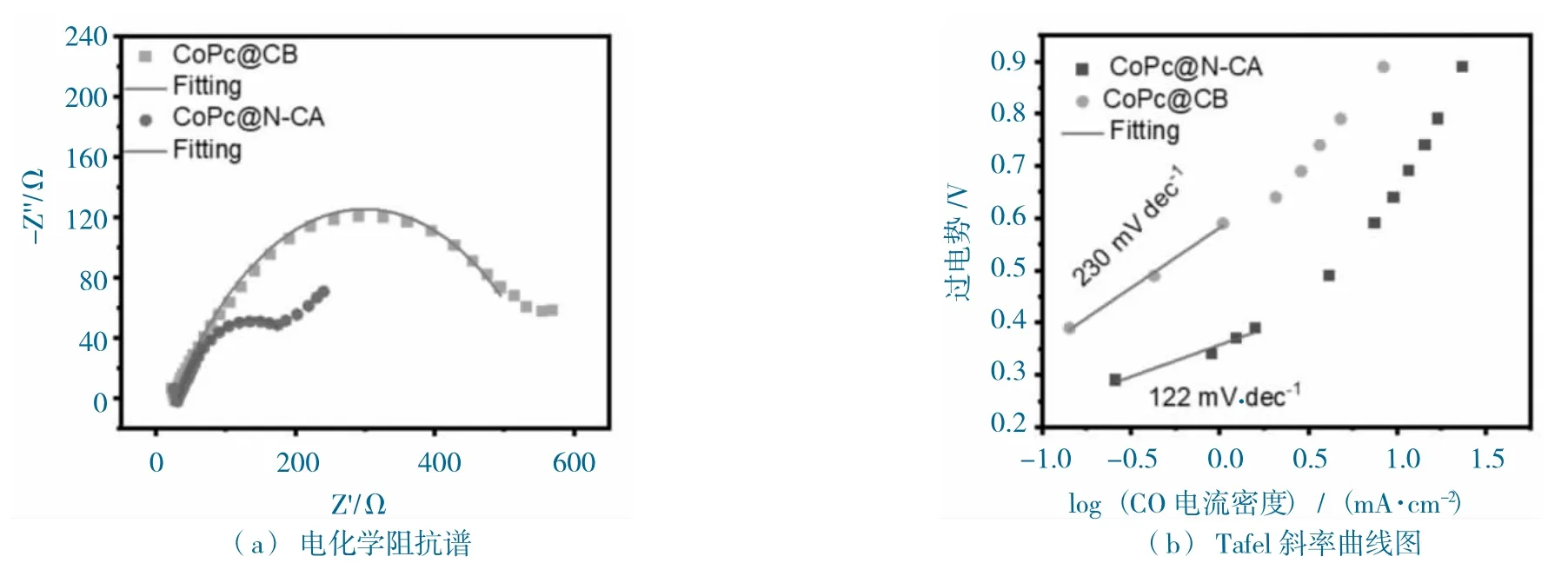
图11 CoPc@N-CA 和CoPc@CB 的阻抗和Tafel 斜率图Fig.11 EIS and Tafel slope diagram of CoPc@N-CA and CoPc@CB
进一步测试了催化剂的动力学性能,EIS 阻抗测试(图11a)发现,CoPc@N-CA 拥有比CoPc@CB更小的半圆,说明CoPc@N-CA 的阻抗值比CoPc@CB 的要小,CoPc@N-CA 拥有更快的电子传导率,这与N-CA 本身是良导体的性能相关。同时,通过催化剂的jCO,推导出CoPc@N-CA 和CoPc@CB 的Tafel 值分别为122 mV·dec-1和230 mV·dec-1(图11b),说明CoPc@N-CA 拥有更快的CO2RR 动力学,这与基底特殊的多孔结构是分不开的。总的来说,CoPc@N-CA 拥有特殊的3D 多孔“类珊瑚状”结构,有利于活性位点的暴露,促进CO2传质,从而增强了CO2RR 的活性和选择性,性能显示优于CoPc@CB。
4 结语
通过传统的水热合成策略制备3D 多孔“类珊瑚状”结构的N-CA,并通过非共价键固定技术,将CoPc 稳定于N-CA 的表面,形成CoPc@N-CA催化剂。基础表征测试证明CoPc 均匀负载于N-CA 的表面,形成均一的活性位点。CoPc@N-CA催化剂中特殊的多孔结构,促进了位点的暴露和CO2的传质,以及N-CA 本身良导体的特质加快了CO2RR 的反应动力学,因此,该催化剂表现出对CO2分子良好的电化学还原性能。在较宽的电势窗口-0.5~-1.0 V vs.RHE 下,CoPc@N-CA 对CO 的选择性>80%,最高为99.05%。同时,在-0.85 V vs.RHE 测试电位下,CoPc@N-CA 对CO 的部分电流密度达到14.40 mA·cm-2,约是CoPc@CB(3.66 mA·cm-2)催化剂的4 倍。本工作不仅为多孔碳气凝胶催化剂上设计高活性的Co 位点提供了一种简单有效的策略,同时该研发的催化剂的高催化活性和好的稳定性说明着其在大规模应用方面极具潜力。
猜你喜欢
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19 09:03:04
陶瓷学报(2021年1期)2021-04-13 01:33:02
军事文摘(2020年20期)2020-11-16 00:31:56
中学生数理化·八年级物理人教版(2020年12期)2020-01-01 15:22:24
长春理工大学学报(自然科学版)(2019年4期)2019-09-02 09:18:06
表面工程与再制造(2019年6期)2019-08-24 06:40:08
中学生数理化·八年级物理人教版(2018年12期)2019-01-31 02:38:18
新型建筑材料(2018年5期)2018-06-14 06:15:02
资源节约与环保(2018年1期)2018-02-08 02:18:27
池州学院学报(2017年3期)2017-10-16 01:38:37
