
PPTA回收溶剂NMP中PPD残留量的测定
2024-01-04 00:13陈建梅苏凤仙
合成纤维工业 2023年6期
陈建梅,苏凤仙*
(1.中国石化仪征化纤有限责任公司研究院,江苏 仪征 211900;2.江苏省高性能纤维重点实验室,江苏 仪征 211900)
聚对苯二甲酰对苯二胺(PPTA)具有高强高模、耐高温、耐腐蚀等多种优良特性,在军工、航天、轮船等领域有着广泛的应用[1-2]。PPTA由对苯二甲酰氯与对苯二胺(PPD)缩聚得到[3],采用N-甲基吡咯烷酮(NMP)为溶剂,每生产1 t PPTA需消耗0.10 ~0.15 t NMP[4],NMP价格昂贵,所以在PPTA聚合过程中需要对NMP进行精馏回收,以降低生产成本。
在PPTA聚合过程中,过量的PPD与聚合异常未反应的PPD会随着溶剂NMP一起流出聚合体系,并参与回收溶剂NMP的分离精馏,但NMP中的PPD无法完全脱除。PPD易被氧化,在空气中能迅速氧化成深褐色,NMP中残留的PPD不仅影响回收溶剂NMP的色相,而且也会干扰聚合粉体PPTA的色相,因此必须严格控制回收溶剂NMP中PPD的残留量。常规PPD的分析方法[5-13]与NMP的分析方法[7-12]不适用于测定NMP中微量PPD的含量。反相高效液相色谱法是一种高效、快速、灵敏的分离分析技术,适合于分离非极性、极性或离子型化合物,对中、高极性物质都有非常高的选择性[14]。
作者采用反相高效液相色谱法测定回收溶剂NMP中PPD的残留量,探讨了高效液相色谱仪的色谱条件,并研究了分析方法的精密度、准确度、检出限,以期为PPTA回收溶剂NMP的质量评定提供参考。
1 实验
1.1 主要试剂及仪器
甲醇:高效液相色谱纯,北京百灵威科技有限公司产;乙腈:高效液相色谱纯,北京百灵威科技有限公司产。
Agilent 1100型高效液相色谱仪:配有可变波长紫外检测器,美国Agilent 公司制;XS105型电子天平:精确至0.1 mg,梅特勒-托利多仪器(上海)有限公司制。
1.2 实验方法
1.2.1 色谱条件
采用等度高效液相色谱法,色谱柱为十八烷基硅烷键合硅胶(ODS)柱(尺寸为4.6 mm×250 mm×5 μm),流动相中水/改性剂(体积比为99:1)与乙腈体积比为95:5,紫外波长为280 nm,进样量为10 μL,定量方法为外标法。
1.2.2 标准曲线绘制
(1)配制一系列不同PPD浓度的NMP溶液,在1.2.1节色谱条件下分别进样分析,得到各浓度对应的峰面积。
(2)以PPD的峰面积为横坐标,浓度为纵坐标绘制标准曲线,如图1所示。
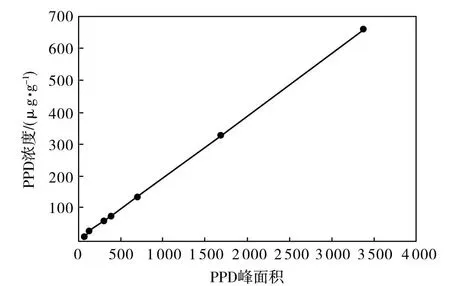
图1 PPD的标准曲线Fig.1 Standard curve of PPD
2 结果与讨论
2.1 色谱条件的选择
2.1.1 色谱柱
非极性烷基键合柱是最广泛使用的液相色谱柱,一般烷基碳链的长度与溶质分离的选择性成正比关系,碳链越长,选择性越好。分别选用八烷基硅烷键合硅胶(C8)柱与ODS柱测定回收溶剂NMP试样中PPD的含量,考察了色谱柱对PPD与NMP组分色谱峰的分离度、PPD组分色谱峰响应值的影响,见图2。
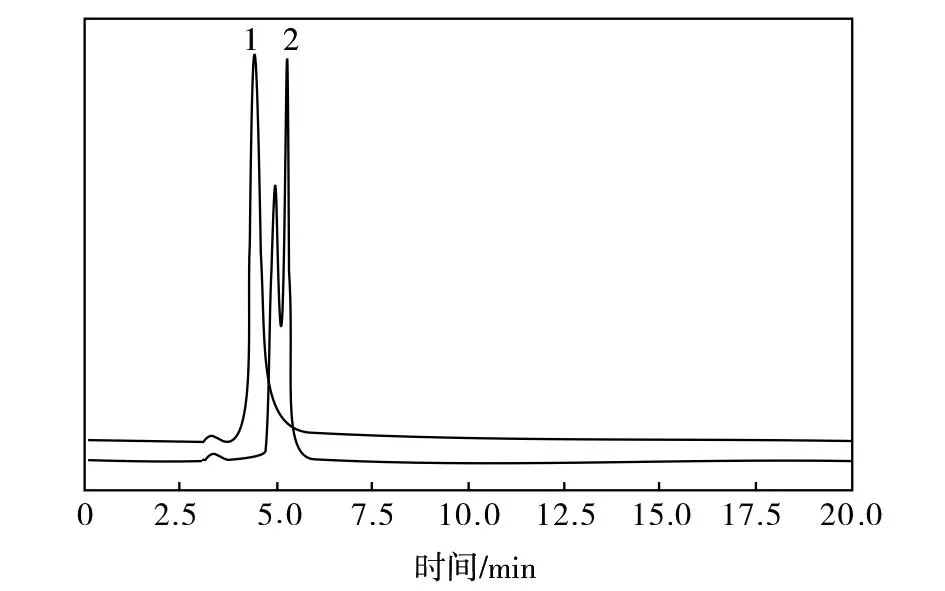
图2 不同色谱柱下回收溶剂NMP的液相色谱图谱Fig.2 Liquid chromatographic spectra of recovered NMP solvent under different chromatographic columns1—ODS柱;2—C8柱
由图2可知,采用ODS柱分析待测试样,PPD组分色谱峰响应值高、峰形对称、无拖尾现象,且与NMP组分色谱峰分离度好,这是因为ODS柱既可在一定程度上增强对弱保留化合物,如苯胺等的保留,也可在一定程度减弱吡咯烷酮类的保留,这对这些化合物的高效快速分离更为有利。
2.1.2 流动相
PPD极性较高,采用反相高效液相色谱法测定时,流动相应选择极性较高的混合溶剂。反相液相色谱的流动相通常以水(二次蒸馏)作为流动相的主体,加入适量乙腈或甲醇改变流动相极性,以此来改善分离选择性。与甲醇相比,乙腈的极性更高,当达到相同强度洗脱能力时,混合溶剂流动相中乙腈配比低于甲醇配比,另外,乙腈用量越小,对环境与实验人员的危害越小,分析成本与能源消耗也越低。
此外,由于待测NMP试样中PPD带有氨基基团,会与色谱柱硅胶填料上的残留硅醇基之间发生相互作用,容易造成离子化,引起PPD色谱峰的拖尾,所以流动相中还需要加入适量改性剂以获得对称的色谱峰。流动相添加改性剂前后回收溶剂NMP的液相色谱见图3。
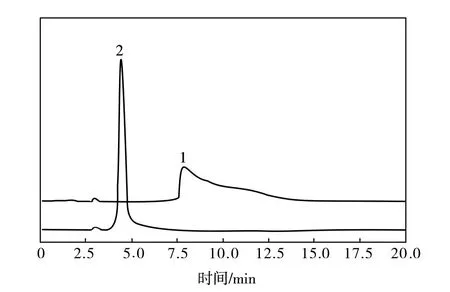
图3 流动相中加入改性剂前后回收溶剂NMP的液相色谱图谱Fig.3 Liquid chromatographic spectra of recovered NMP solvent before and after adding modifiers to mobile phase1—改性剂加入前;2—改性剂加入后
由图3可知,添加改性剂后PPD组分的色谱峰峰形较好,没有拖尾现象。另外,经过实验对比,发现当流动相中水/改性剂(体积比为99:1)与乙腈体积比为95:5时,PPD组分色谱峰分离效果较好,所以选用此比例流动相测定回收溶剂NMP中PPD的残留量。
2.1.3 紫外波长
紫外波长可以影响待测组分的峰响应值、组分分离度、分析时间等,选择合适的紫外波长能提高回收溶剂NMP中微量PPD分析的灵敏度、分离度,使定量结果更准确。NMP的紫外吸收波长为235 nm,PPD的紫外吸收波长为304 nm,为了突出PPD组分,减弱NMP组分色谱峰的干扰,考察了紫外波长对PPD与NMP组分色谱峰分离度、PPD组分色谱峰响应值的影响,见图4。
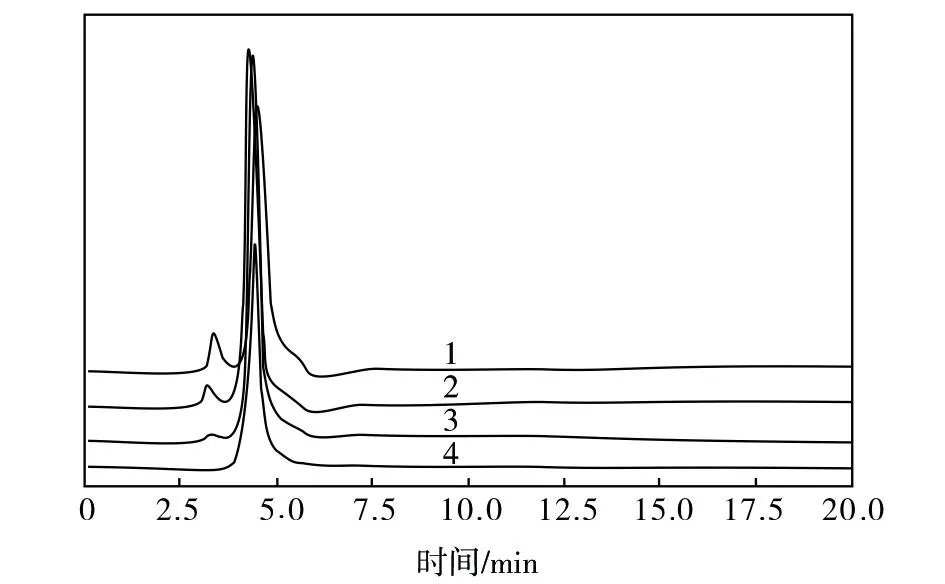
图4 不同紫外波长下回收溶剂NMP的液相色谱图谱Fig.4 Liquid chromatographic spectra of recovered NMP solvent at different ultraviolet wavelengths1—240 nm;2—260 nm;3—280 nm;4—300 nm
由图4可知:紫外波长为240 nm、260 nm时,PPD组分色谱峰峰形拖尾较严重,不利于准确定量;紫外波长为280 nm时PPD与NMP组分色谱峰分离度较好,且NMP组分色谱峰响应值较低,说明此紫外波长条件下可以避免NMP的干扰,突出PPD组分色谱峰,有利于PPD的准确分析,所以选择紫外波长为280 nm测定回收溶剂NMP中PPD的残留量。
2.1.4 流动相流速(u)
由范第姆特公式(见式1[14])可知,u的增大或减小,对柱效(以理论塔板高度(H)来表示,H越小,柱效越高)没有绝对的影响,当u增大时会增大传质阻力项,使H增大、柱效降低,但u增大同时会减小分子扩散项,使H减小、柱效增大,所以考察了u对PPD与NMP组分色谱峰分离度、PPD组分色谱峰响应值的影响。
(1)
式中:A为涡流扩散项,B为分子扩散系数,C为传质阻力系数。
由图5可知,当u为1.0 mL/min与1.2 mL/min时,PPD组分色谱峰峰形尖锐,无拖尾现象,并且与NMP组分色谱峰的分离度较好。考虑到液相色谱分析中常用的u为1.0 mL/min,所以选择u为1.0 mL/min测定回收溶剂NMP中PPD的残留量。
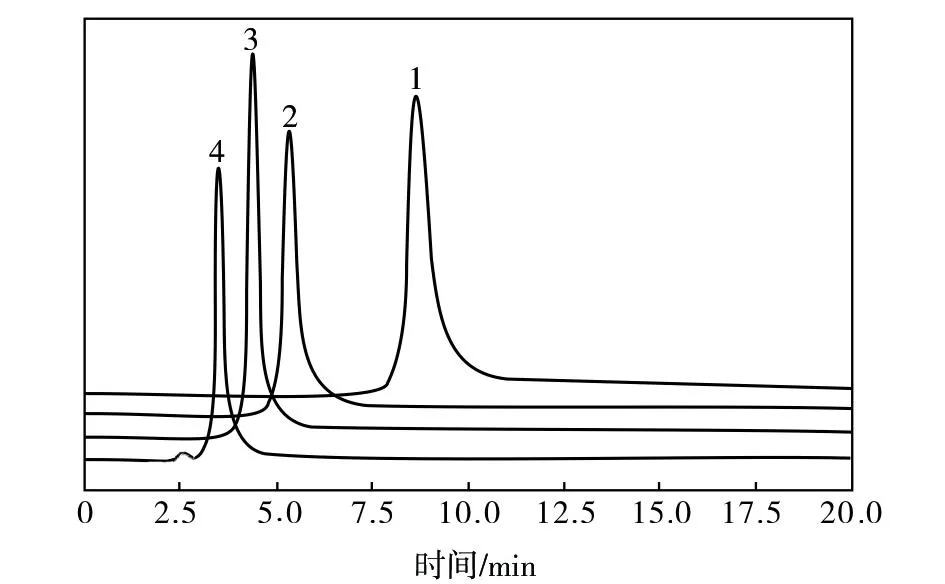
图5 不同u下回收溶剂NMP的液相色谱图谱Fig.5 Liquid chromatographic spectra of recovered NMP solvent at different u1—0.5mL/min;2—0.8 mL/min;3—1.0 mL/min;4—1.2 mL/min
2.2 试样分析
配制PPD含量为10~400 μg/g的NMP溶液,按照1.2.1节色谱条件,考察了回收溶剂NMP中PPD含量对PPD与NMP组分色谱峰分离度、PPD组分色谱峰响应值的影响,见图6。
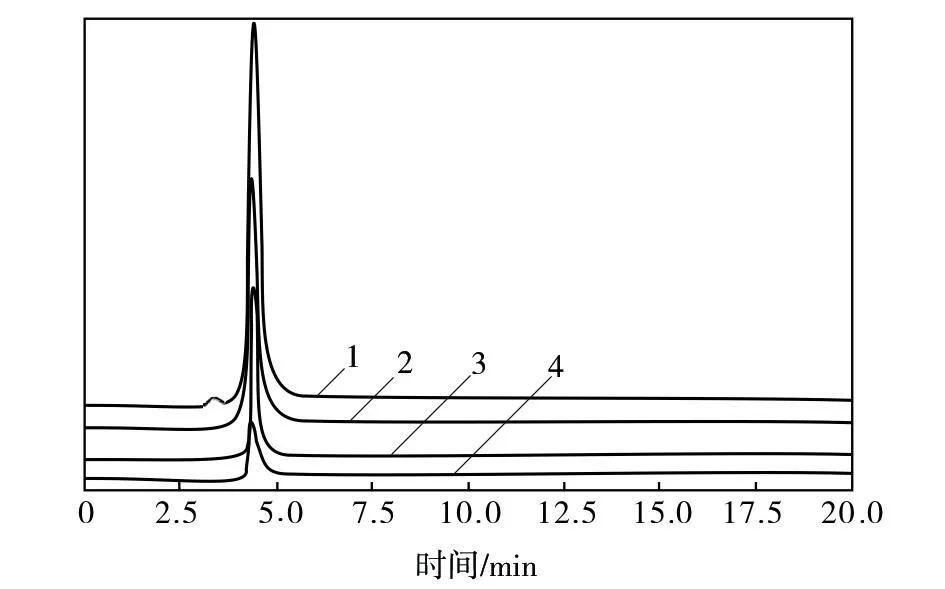
图6 不同PPD含量回收溶剂NMP的液相色谱图谱Fig.6 Liquid chromatographic spectra of recovered NMP solvent at different PPD content1—400 μg/g ;2—200 μg/g;3—100 μg/g;4—10 μg/g
由图6可知,回收溶剂NMP中PPD含量为10~400 μg/g时,PPD组分的色谱峰响应值均非常高,峰形对称,无拖尾现象与峰展宽现象,且NMP组分色谱峰的响应值非常低,可以排除NMP色谱峰对PPD定量的干扰,这说明高效液相色谱法的色谱条件较好,适用于回收溶剂NMP中微量PPD含量的测定。
2.3 精密度
按照1.2.1节色谱条件对回收溶剂NMP试样中PPD含量进行分析,平行测定6次,结果见表1。由表1可知,该方法的相对标准偏差(RSD)为0.85%,低于1%,说明采用反相高效液相色谱法测定回收溶剂NMP中PPD含量的精密度误差小,满足分析测试要求。
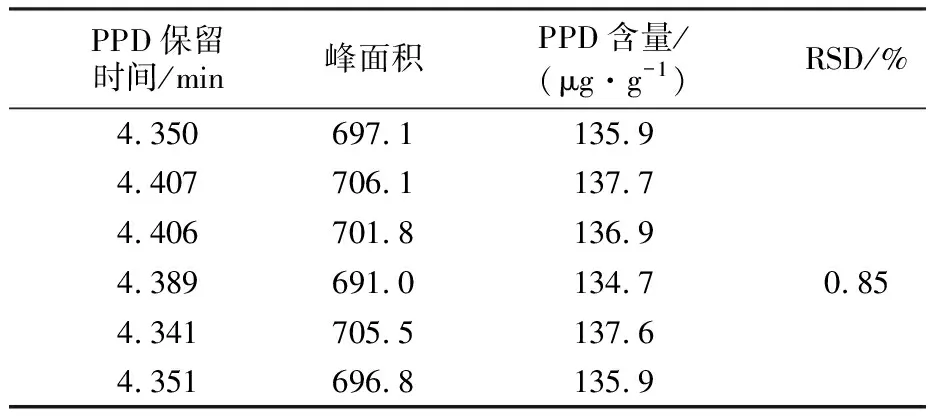
表1 方法的精密度实验结果Tab.1 Precision experimental results of determination method
2.4 准确度
在回收溶剂NMP中添加不同质量的PPD,溶解后按照1.2.1节色谱条件测定各溶液中PPD的含量,再以式(2)计算加标回收率(K),结果见表2。
(2)
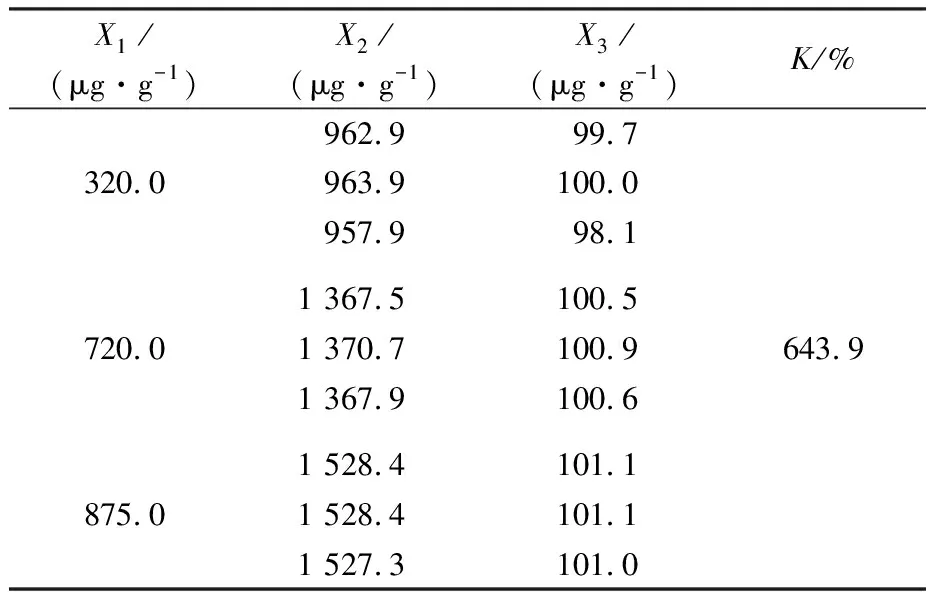
表2 方法的准确度实验结果Tab.2 Accuracy experimental results of determination method
式中:X1为试样实际量,X2为试样添加量,X3为试样测定值。
从表2可以看出,该方法的K为98.1%~101.1%,说明该方法的准确度较优。
2.5 线性范围与检出限
线性范围指响应值(或峰面积)随组分浓度变化曲线上直线部分所对应的组分浓度变化范围。在色谱分析中一般将检出限(D)定义为信噪比(S/N)为3:1时的浓度[15],以式(3)计算。
(3)
式中:Q为进样浓度。
按照1.2.1节色谱条件,以式(3)计算反相高效液相色谱法测定回收溶剂NMP中PPD含量的D,结果列于表3。由表3可知:该方法具有较宽的线性范围和良好的线性关系,相关系数为0.999;该方法也具有较高的灵敏度,D为0.35 μg/g,满足PPTA回收工艺的分析要求。

表3 方法的线性范围与DTab.3 Linear range and D of determination method
3 结论
a.采用反相高效液相色谱法测定PPTA回收溶剂NMP中微量PPD的含量,适宜的色谱条件为色谱柱采用十八烷基键合硅胶(ODS)柱、尺寸为4.6 mm×250 mm×5 μm,流动相中水/改性剂(体积比为99:1)与乙腈体积比为95:5,波长为280 nm,进样量为10 μL,定量方法为外标法。
b.该分析方法精密度优、重复性好、线性方程的相关性好,RSD为0.85%,K为98.1%~101.1%,D为0.35 μg/g,能满足PPTA回收溶剂NMP中微量PPD含量的测定。
猜你喜欢
煤化工(2022年3期)2022-07-08
科教导刊·电子版(2021年1期)2021-03-28
山东交通科技(2020年1期)2020-07-24
环境与发展(2019年11期)2019-02-12
山东化工(2019年1期)2019-01-24
中国资源综合利用(2016年10期)2016-01-22
中国塑料(2015年6期)2015-11-13
中国塑料(2015年10期)2015-10-14
丝绸(2014年2期)2014-02-28
重庆理工大学学报(自然科学)(2012年12期)2012-09-18
