
均相合成多重选择性的新型两亲性C22高效液相色谱固定相
2024-01-02 10:43:36范二乐蒋星宇张加栋张明亮韩海峰张大兵
分析测试学报 2023年12期
范二乐,蒋星宇,张加栋*,张明亮,韩海峰,,张大兵,,陈 义,3
(1.淮阴工学院 矿盐资源深度利用技术国家地方联合工程研究中心,高端矿盐功能材料智能制备国际合作联合实验室,江苏 淮安 223003;2.江苏汉邦科技股份有限公司,江苏 淮安 223000;3.中国科学院化学研究所活体分析化学科学院重点实验室,北京 100190)
色谱固定相的性质决定了保留机理、分离效率以及适合的分离对象[1-2]。反相液相色谱(RPLC)是目前最为常用的一类分离模式,有丁基(C4)、辛基(C8)、十二烷基(C12)、十八烷基(C18)等各种疏水性固定相[3-4]可供选择。但RPLC 不适用于对极性特别是强极性化合物的分离。在其烷基链上嵌入极性基团,如脲(U)[5-7]、氨基甲酸酯[8-9]、酰胺(A)[10-14]、咪唑啉[15]、磷酸根[16]、季铵[17]等基团是改善其对极性化合物分离性能的一种有效手段。嵌入基团还能改变或引入新的选择机制,比如酰胺基会引入氢键等新作用机制,因而可以成为一种多模式或混合模式分离的新型高效液相色谱(HPLC)固定相[7]。
在嵌入型固定相的研究方面,Nogueira 等[18]合成了嵌有硫醚基团的十一烷基氨基奎尼考定固定相;Qiu等[19]制备了聚1-(2-丙烯酰氧十二烷基)-3-甲基咪唑溴化铵固定相;Mallik 等[20]制得了嵌有多重酰胺的C18 固定相等。上述研究将多个基团引入色谱固定相主要存在以下问题:如功能分子的键合过程常在非均相反应状态下进行,易出现固定相密度波动等问题,给色谱行为考察和结果解读等造成一定的困难;此外,非均相反应需要较繁复的基团保护与脱保护过程,对选择嵌入目标基团非常不利,当仲胺和伯胺共存时,若不用叔丁氧酰基进行多次保护,很难选择嵌入到对称多胺化合物中[20-21]。
另一方面,在均相反应条件下,基团可实现选择性功能化,从而有效避免上述情况的出现。比如,对于酰胺的合成,酯在特定催化剂作用下可以实现多胺的选择性酰化[22]。同时,N-酰基咪唑也可以高选择性、高效地酰化伯胺,生成酰胺,省去了繁琐的保护和脱保护过程,且反应条件温和[23]。
本文利用均相反应合成两种新型两亲性色谱固定相。首先,在催化剂的作用下,选择性酰化合成了N-[3-(三甲氧基硅)丙基-N′-二十二酰乙二胺(C22-A-硅烷);然后,利用免保护酰胺化反应将酰胺外的仲胺转化成脲基,制备了酰胺/脲双嵌入的N-[3-(三甲氧基硅)丙基-N-二甲胺甲酰基-N′-二十二酰乙二胺(C22-A/U-硅烷);最后,经硅烷化反应将上述两种硅烷键合到硅胶载体上,得到目标固定相C22-A 和C22-A/U。相较于非均相接枝法合成的硅胶固定相,这种均相合成目标硅烷并键合至硅胶载体的方法免除了繁复的基团保护与脱保护过程,具有更高的转化率,可保证接枝体的均一性,非常有助于固定相色谱行为的解读。实验结果表明,两种新型硅胶固定相既能用于反相液相色谱分离也能用于亲水相互作用色谱分离,展现出非常好的两亲性能,具有一定的应用开发前景和理论研究价值。
1 实验部分
1.1 试剂与仪器
全多孔球形硅胶(平均直径:5 µm,比表面积:350 m2/g,孔容:1.0 mL/g)购于日本AGC 公司;C18 改性色谱硅胶(粒径:5 µm)购于日本富士公司;色谱柱均采用4.6 mm×150 mm 的不锈钢柱装填;二十二酸、N,N′-羰基二咪唑(CDI)、六甲基二硅氮烷(HMDS)、N,N-二甲氨基甲酰氯、雌激素等购于上海安耐吉化学有限公司;N-[3-(三甲氧基硅基)丙基]乙二胺(AEAPS)购于上海阿拉丁生化科技股份有限公司;色谱级甲苯(Tol)、甲醇(MeOH)、乙腈(MeCN)均购于国药化学试剂有限公司;标准参照物(SRM 870,SRM869b)购于美国国家标准技术研究所(NIST)。扫描电子显微镜(SEM)为S-3000N(日本HITACHI 公司),固态13C 核磁共振仪为JNM-ECZ 600R(日本电子株式会社(JEOL)),元素分析由ElementarVario EL III型元素分析仪(德国哈瑙)完成。
1.2 固定相的制备
1.2.1 C22-A 硅胶键合固定相的制备称取50 mmol 二十二酸(乙酸乙酯重结晶3 次)溶于50 mL 甲苯;共沸除水,待降温至90 ℃后,少量多次地加入50 mmol 的CDI,反应2 h;再加入N-[3-(三甲氧基硅基)丙基]乙二胺,继续反应8 h;降温至70 ℃,减压蒸馏除去甲苯后,加入75 mL乙腈,充分溶解后静置冷却结晶。产物C22-A-硅烷经离心收集,用乙腈洗涤2次,干燥后待用。
称取10 g 硅胶,在机械搅拌下悬浮于50 mL 甲苯中;加入7 g 干燥的C22-A-硅烷,回流反应24 h;趁热过滤,用热甲苯洗涤3 次,70 ℃烘干。随后悬浮于甲苯中,用六甲基二硅氮烷做封尾处理20 h。反应结束后过滤收集C22-A 键合硅胶颗粒,用沸甲醇洗涤3 次,烘干后即可用于装柱。流程如图1 中Route 1所示。

图1 C22-A与C22-A/U键合硅胶固定相的制备路径Fig.1 Routes to prepare on-silica-bonded C22-A and C22-A/U stationary phases
1.2.2 C22-A/U 硅胶键合固定相制备取12 g 干燥C22-A-硅烷溶于甲苯,加入等摩尔三乙胺;在70 ℃下滴加等摩尔N,N-二甲氨基甲酰氯,反应4 h。过滤除去三乙胺的盐酸盐,取滤液蒸出甲苯,再用无水乙腈洗涤后与硅胶键合,135 ℃回流24 h,其余处理同上。制备流程如图1中Route 2所示。
1.3 色谱柱制备
色谱柱由匀浆液填充法制备。称取2.2 g C22-A或C22-U/A 键合硅胶颗粒,悬浮于40 mL三氯甲烷中,超声匀浆。在50 MPa液压下,以乙腈为顶替液,将匀浆压入不锈钢柱管(4.6 mm×150 mm)中。装填完成后,将色谱柱安装至液相色谱仪上,用纯甲醇冲洗30 min后备用。
1.4 色谱分析
所有色谱测试均在汉邦Newstyle 高效液相色谱系统上进行。该装置配备NP 7000 二元泵、NU 3000紫外-可见检测器、柱温箱和Rheodyne 7725进样器。采用等度洗脱分离,参数设定为:流速1.0 mL/min、柱温30 ℃、进样量5 µL、紫外检测波长254 nm。所有流动相和分析样品在使用前均经0.45 µm多孔膜过滤,临用时再超声脱气。
RPLC:用4∶1(体积比)的甲醇/20 mmol·L-1中性磷酸钾缓冲液分离SRM870,用9∶1(体积比)的甲醇/水分离烷基苯、烷基联苯、三联苯异构体、三亚苯,用纯甲醇分离SRM869b。
HILIC:用体积比为5∶95~95∶5 的乙腈/20 mmol·L-1乙酸铵缓冲液分离甲苯与胞苷,用纯水分离胞苷与尿苷,用15∶85(体积比)的乙腈/0.1%甲酸水溶液分离磺胺类药物,用3∶2的甲醇/水分离三环抗抑郁药,用3∶1的甲醇/水分离雌激素(在230 nm检测)。
2 结果与讨论
2.1 色谱固定相表征
2.1.1 元素分析及色谱性能表征表1 汇总了新制备的C22-A、C22-A/U 与传统C18 固定相的重要参数。表1 显示,C22-A 和C22-A/U 键合硅胶固定相比商品C18 固定相多出了氮元素,且C22-A/U 的相对含氮量(1.83%)约为C22-A(1.38%)的1.5倍,初步说明含氮基团(酰胺和脲基极性基团)的存在,碳、氮元素同时出现且比例符合理论值,说明预先合成的功能化硅烷均已成功键合到硅胶上。C22-A/U 和C22-A 的区别在于前者的键合密度(2.18 µmol·m-2)高于后者(1.89 µmol·m-2),脲基对键合量的提升具有促进作用。C22-A 和C22-A/U 的键合密度均小于C18(2.67 µmol·m-2),原因可能是二者的二十二烷基碳链更长,位阻更大。
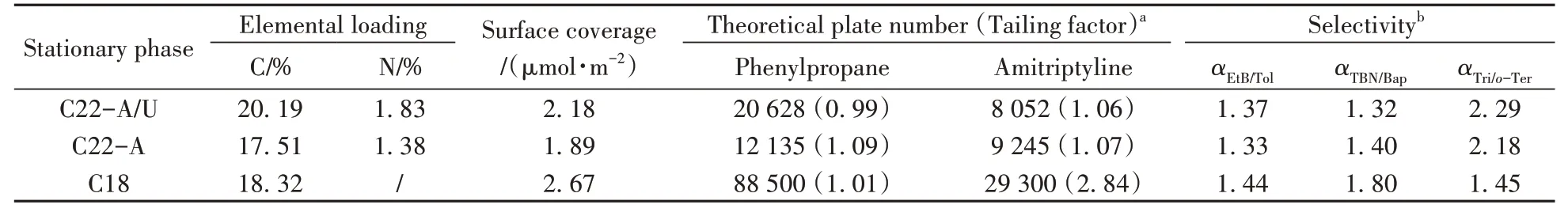
表1 C22-A、C22-A/U键合硅胶固定相的元素分析与色谱性能表征结果Table 1 Elemental analysis and chromatographic characterization of C22-A and C22-A/U bonded silica gel
2.1.2 固态13C 核磁共振波谱分析为进一步证明硅烷配体的结构,采用固态13C 核磁共振技术表征了新制备的C22-A固定相(图2)。其中,化学位移174.1 ppm处为酰胺基的羰基碳振动峰,30 ppm处为亚甲基碳的主峰,对应于长脂肪链的扭曲构象,此构象是可移动和无定形的[24];而30.7 ppm 处的小肩峰证明了反式构象的存在,此构象是刚性和有序的[25],但反式构象不明显。由此可见C22 的大部分链处于无序状态,其原因可能是键合密度较低。结果表明含酰胺极性配体的硅烷成功接枝到硅胶表面,同时证实了配体结构。
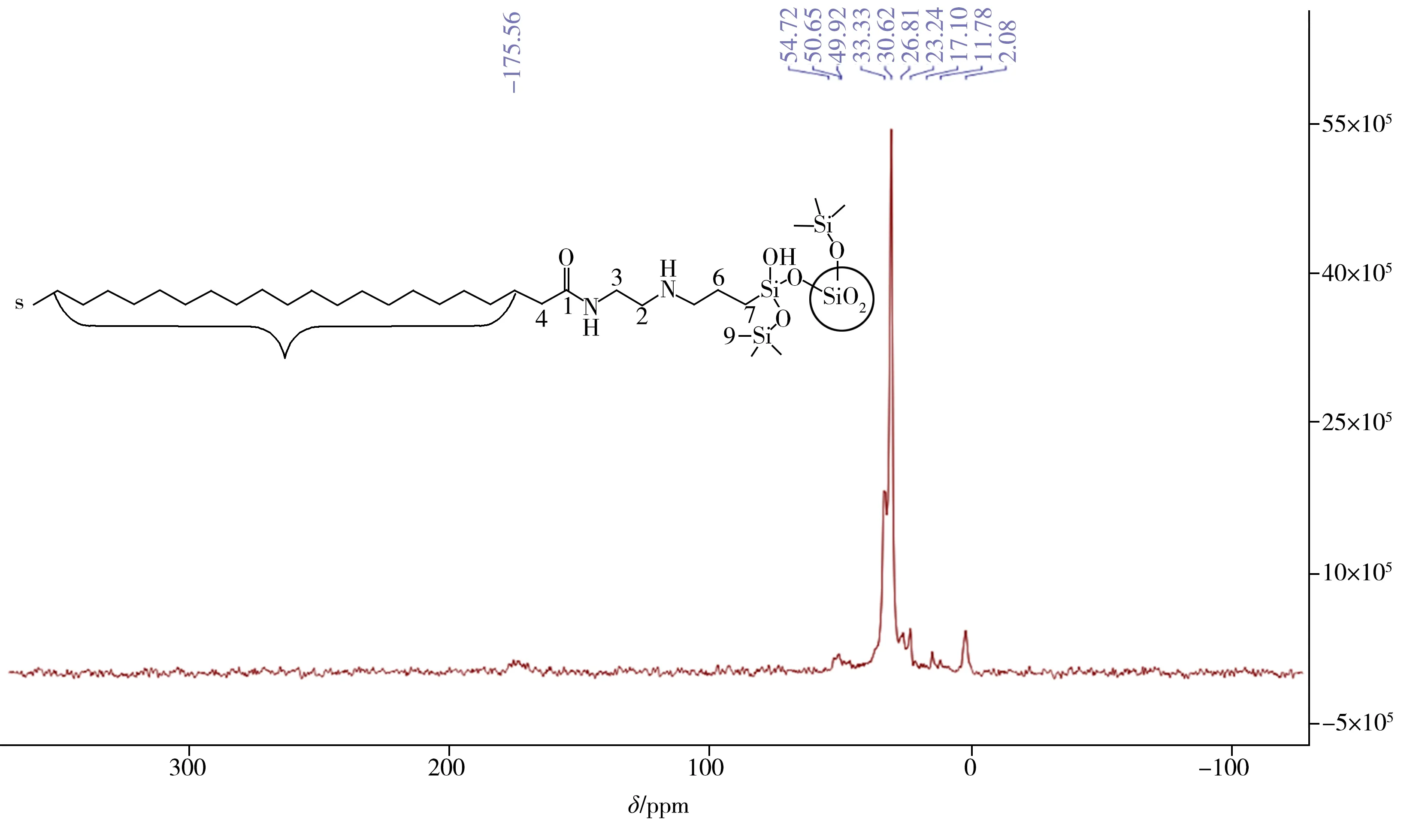
图2 C22-A固定相的固态13C核磁共振谱图Fig.2 Solid-state 13C NMR spectrum of C22-A
C22-A/U 固定相的固态13C 核磁共振波谱如图3 所示,175 ppm 处与30 ppm 处的峰信号明显,分别为酰胺和脲基的羰基碳的吸收峰以及碳二十二链的亚甲基碳的吸收峰,说明极性基团和碳二十二链成功键合到硅胶表面。
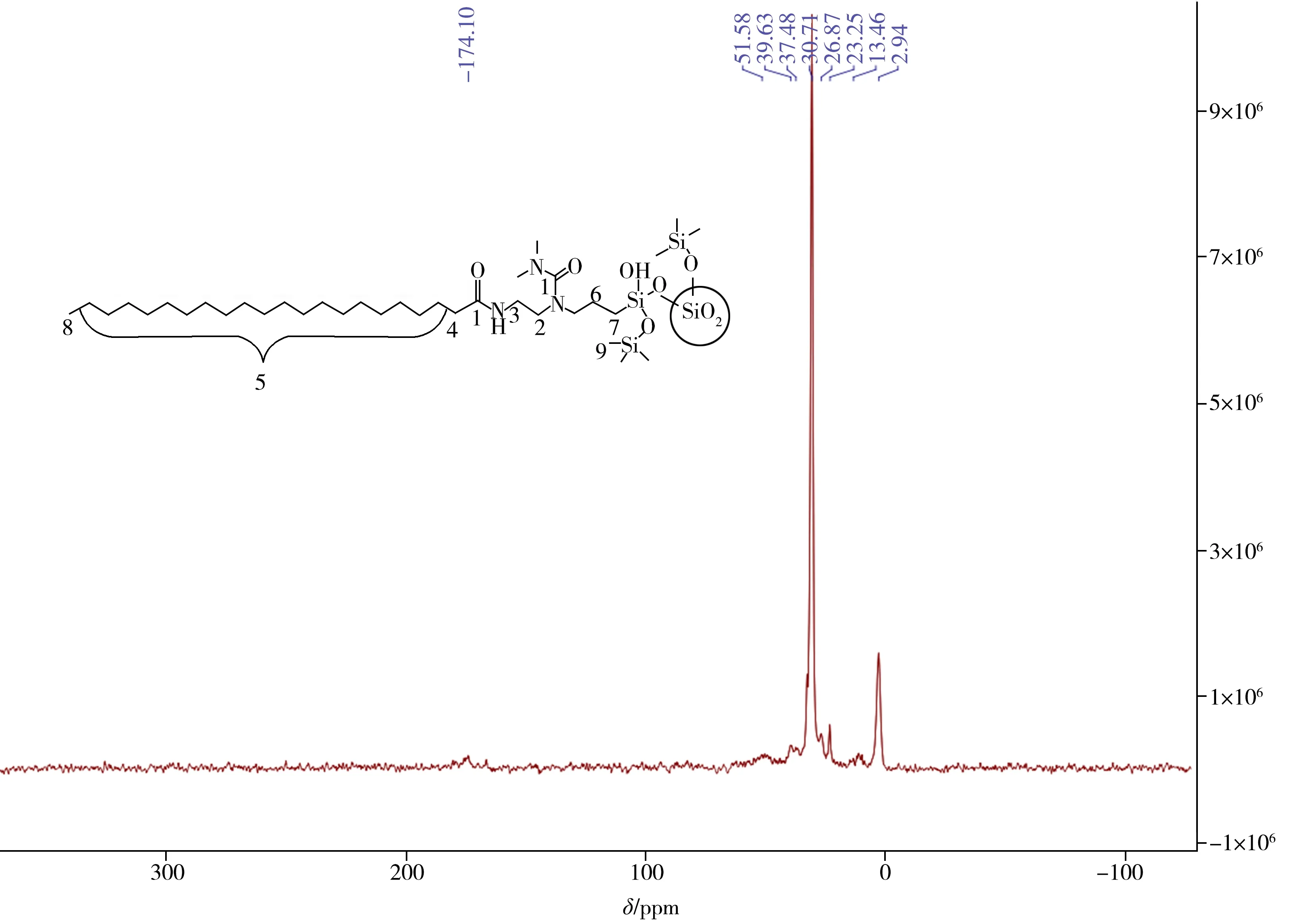
图3 C22-A/U固定相的固态13C核磁共振谱图Fig.3 Solid-state 13C NMR spectrum of C22-A/U
2.1.3 扫描电子显微镜表征对未键合硅胶和C22-A 和C22-A/U 的硅胶固定相分别进行扫描电子显微镜(SEM)表征,结果如图4 所示。从SEM 图可以看出,硅胶键合前后的外观形貌无明显变化,硅胶微球未发生粘连、破裂等不良情况,微球形状规则、单分散性良好。由此可以证明,硅胶微球的机械强度和分离稳定性良好,不易发生恶性形变,适于色谱柱的装填。

图4 裸硅胶(A)、C22-A(B)以及 C22-A/U(C)的SEM图Fig.4 SEM images of bare silica gel(A),C22-A(B) and C22-A/U(C)
2.2 色谱固定相性能分析
2.2.1 残余硅羟基活性与疏水选择性色谱固定相多数键合在硅胶载体上,常会残留硅羟基等非特异性吸附活性位点,易导致峰拖尾、柱效下降等问题。合成的新型固定相在靠近硅胶表面一端碳链上嵌有脲与酰胺基团,链之间在此处易形成氢键,可能形成覆盖活性位点的大网络,从而可以有效屏蔽活性位点产生的不利影响;同时嵌入基团也可能影响固定相的疏水选择性和芳香选择性。为此,采用仿SRM870[26]五组分配方进行色谱表征:其中,以尿嘧啶测量死时间,以碱性阿米替林考察硅胶表面残余硅羟基的活性,以甲苯和乙苯测定疏水选择因子。
如图5 所示,阿米替林在C18 柱上拖尾明显,保留时间长(图5A),而在C22-A 和C22-A/U 上出峰对称且快速(图5B、C),表明嵌入的酰胺和脲基能有效屏蔽硅羟基的不利作用。如表1 所示,相较于阿米替林在常规C18 固定相的拖尾因子(2.84),其在C22-A 和C22-A/U 的拖尾因子分别为1.07 和1.06,表明双重嵌入的极性基团极大地降低了固定相硅羟基的活性,为碱性化合物的分离奠定了基础。
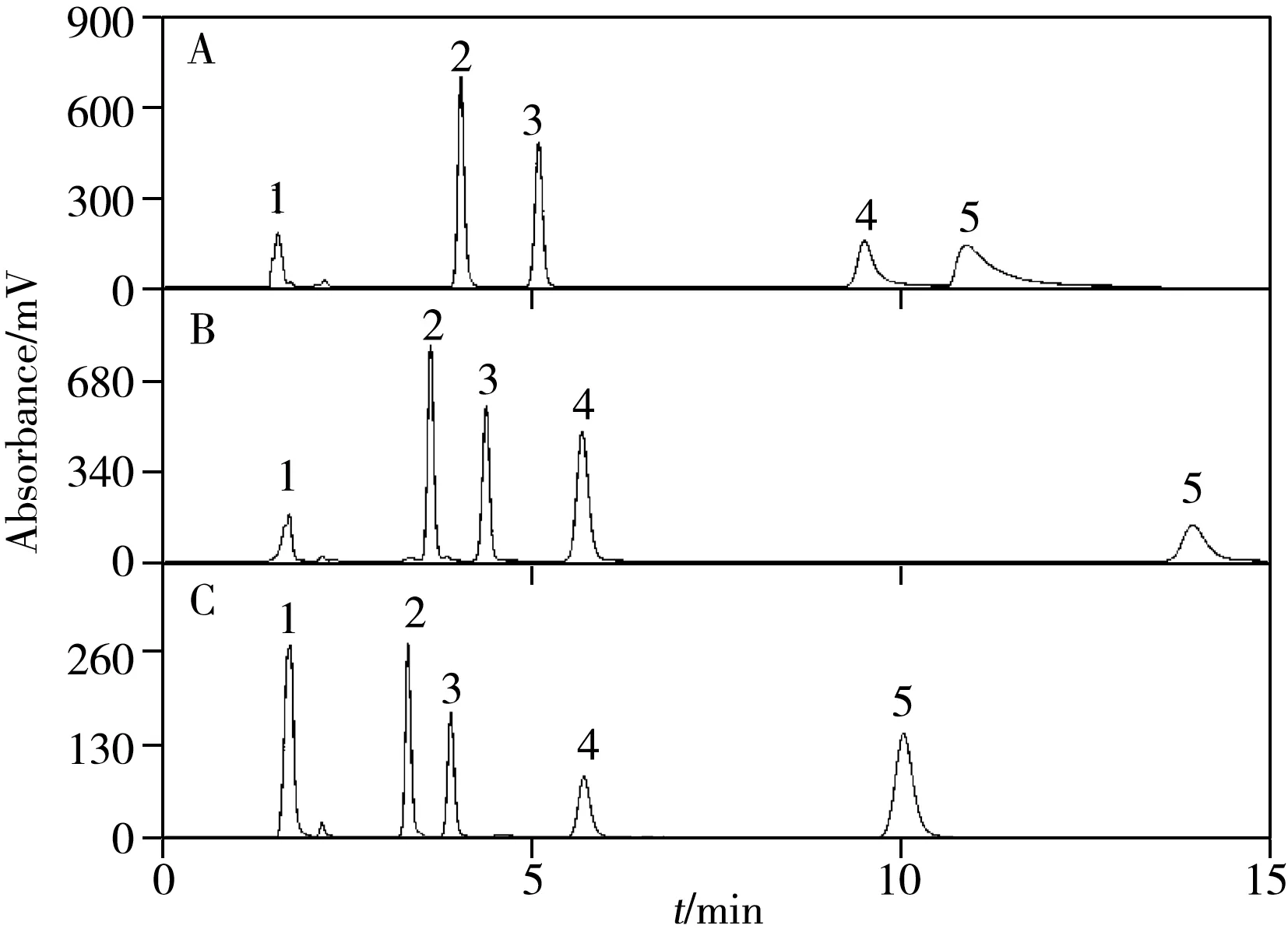
图5 自配仿SRM870在 C18(A)、C22-A/U(B)和C22-A(C) 柱上测得的色谱图Fig.5 Chromatograms of laboratory-prepared SRM870 eluted by C18(A),C22-A/U(B) and C22-A(C) columns
由表1 可知,C22-A 和C22-A/U 由于嵌入了亲水基团会抵消一些碳链的疏水作用,柱效比应用于RPLC的C18差,αEtB/Tol也稍低于C18,但足以基线分离甲苯和乙苯,初步表明利用该方法制备的两种极性嵌入型固定相可以用于RPLC分离。
2.2.2 形状选择性两种新固定相还具备形状分辨能力,可用多环芳烃混合物SRM869b[27-28]的色谱分离(图6)来检验。SRM869b 的选择因子(αTBN/BaP)小于1.0 时为强形状选择性,大于1.7 时为弱选择性,介于1.0~1.7 时为中等强度选择性。C22-A/U 和C22-A 对四苯并芘和苯并芘的选择因子介于1.0~1.7 之间(分别为1.32、1.40),为中等强度选择性;而C18 测得值为1.80,形状选择性更弱。
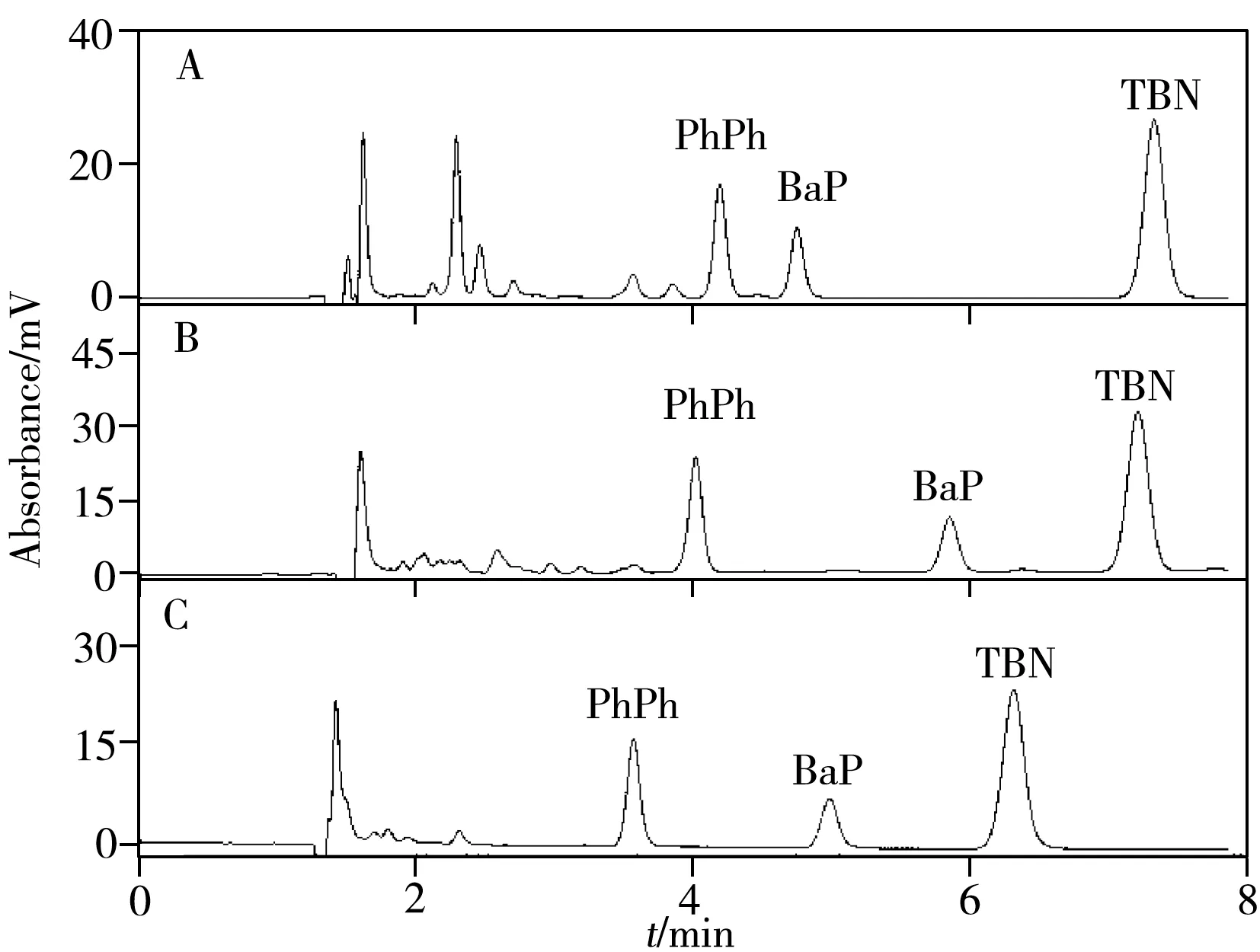
图6 用 C18(A)、C22-A/U(B)和C22-A(C)柱测得的SRM869b色谱图Fig.6 Chromatograms of SRM869b eluted by C18(A),C22-A/U(B) and C22-A(C) columns
以邻三联苯(o-Ter)、间三联苯(m-Ter)、对三联苯(p-Ter)、三亚苯(Tri)为样品,分析了新固定相对平面异构体的识别能力,其中Tri 和o-Ter 的长宽比接近,但分子平面结构不同,其αTri/o-Ter可度量固定相的平面选择性[29],结果是:C22-A/U(2.29)>C22-A(2.18)>>C18(1.45)。这说明带有酰胺和脲两种羰基极性基团的C22 烷基链能强化固定相的平面识别能力。C22-A 对多环芳烃异构体良好的形状识别能力,是因为固定相与多环芳烃之间存在羰基π-苯π 相互作用,而脲基与酰胺结构相似,结果显示嵌有酰胺、脲基双重基团的固定相的形状选择性略优于单酰胺的C22-A。
2.2.3 亲水作用分析C22-A 和C22-A/U 嵌有强极性亲水基团,具有兼容100%水为流动相的可能性。以纯水为流动相分离了胞苷和尿苷。在纯水流动相中,多次重复进样获得的保留时间(表2)不随进样次数缩短,说明二者均具有可被水完全浸润的特征[7]。

表2 由C22-A和C22-A/U测得的胞苷和尿苷的保留时间变化Table 2 Retention time changes of cytidine and uridine measured by C22-A and C22-A/U
以甲苯和胞苷为基准物,在HILIC模式下测得了两者的保留因子随流动相中乙腈比例变化的曲线。图7A显示,在C22-A上胞苷的保留因子只在很窄的范围内随乙腈体积分数的增加而增强,可见C22-A表现出典型的HILIC 保留机制,不具备混合色谱模式特征。对于C22-A/U(图7B),甲苯的保留因子随乙腈体积分数的下降而上升,是典型的RPLC 特征;而胞苷的保留因子则先降后升,呈现U 型曲线,具有混合模式保留特征。通过调节流动相的组成和配比[30],可实现C22-A 和C22-A/U 在不同模式下的分离,极大地方便了操作。
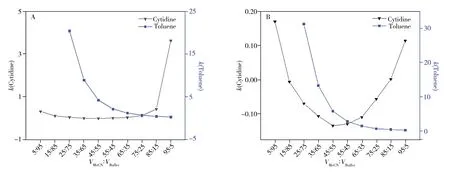
图7 在C22-A(A)和 C22-A/U(B)上测得的甲苯与胞苷保留因子随流动相中乙腈体积分数的变化曲线Fig.7 Retention factor plots of toluene and cytidine against volume fraction of acetonitrile in the mobile phases measured on C22-A(A) and C22-A/U(B) columns
2.3 新型色谱固定相应用
2.3.1 碱性化合物的分离为考察新制备固定相对碱性化合物的分离能力,以阿米替林等7种三环抗抑郁药组成的混合物作为分析物对两种色谱柱进行了评价(图8),7种碱性化合物40 min内在两种色谱柱上全部被洗脱,且峰形较好。C22-A/U 对阿莫沙平和多塞平表现出更优越的分离能力,这也体现了嵌入脲基的优势,出现此现象的原因还有可能是C22-A/U 的含碳量高于C22-A。总体来说,此两种固定相对碱性化合物具有良好的识别能力,嵌入极性酰胺基和脲基的C22 固定相在一定程度上可以解决传统C18固定相对碱性化合物分离拖尾严重或保留不足的问题。
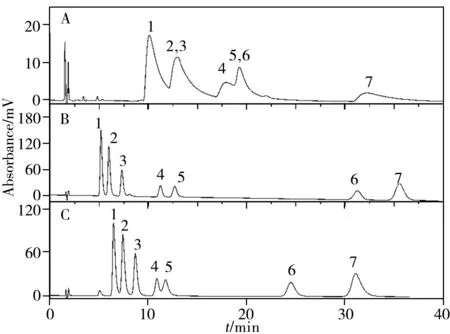
图8 7种三环抗抑郁药在C18柱(A)、C22-A/U柱(B)和C22-A柱(C)上的色谱图Fig.8 Chromatograms of 7 types of tricyclic antidepressants on C18(A),C22-A/U(B) and C22-A(C) columns
2.3.2 雌醇(酮)类化合物的分离使用C22-A、C22-A/U和C18柱分离含雌三醇、雌酮以及两种雌二醇异构体的四组分溶质(图9),结果表明C22-A和C22-A/U 可以实现4种雌激素的完全分离,而C18未能完全分离,说明两种极性嵌入固定相对雌激素的多环结构表现出良好的亲和力。其中,C22-A/U 对雌酮和雌二醇异构体的选择性远高于C22-A,说明溶质和脲基之间氢键相互作用的重要性。
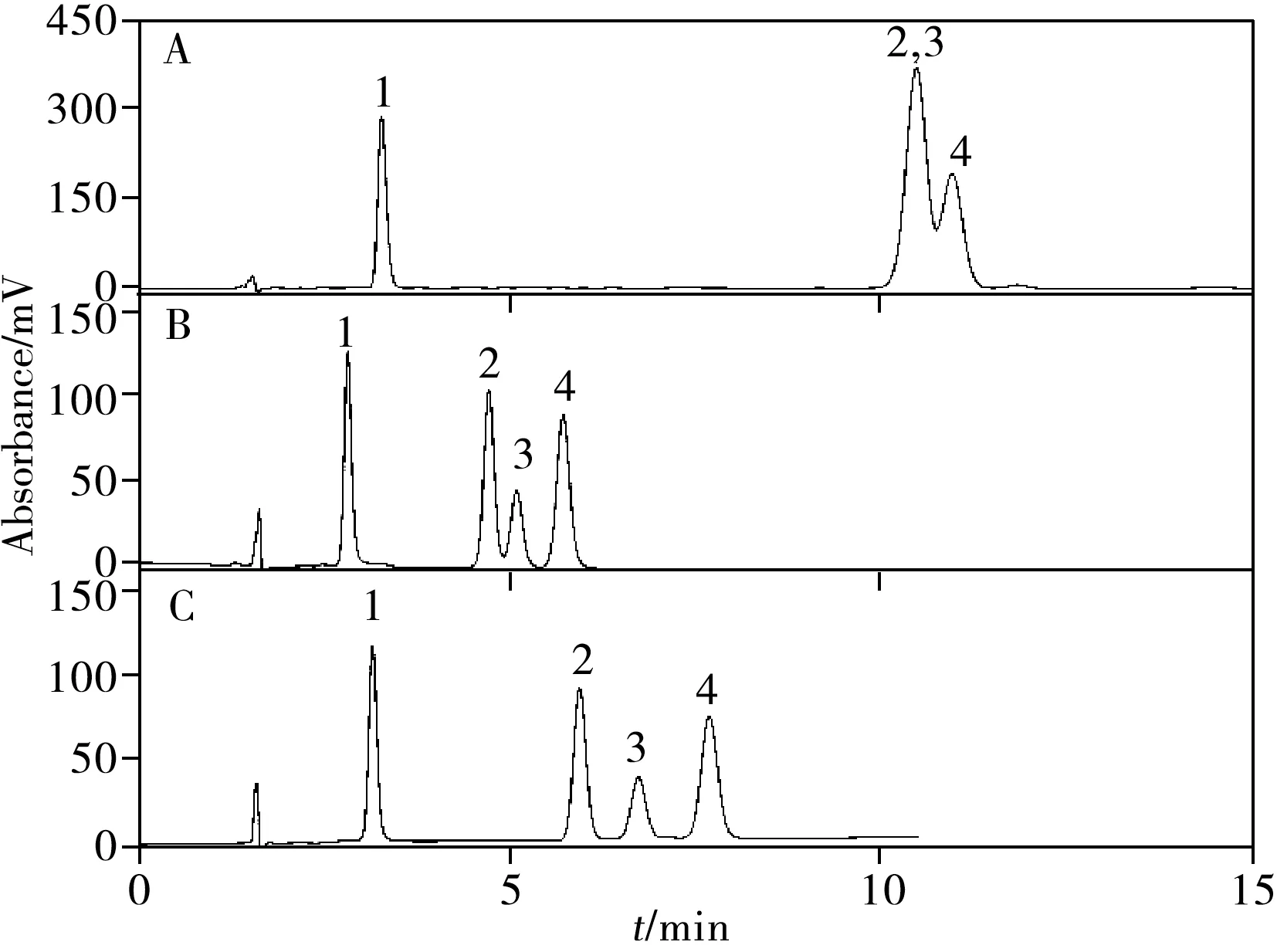
图9 4种雌激素在C18柱(A)、C22-A(B)柱和C22-A/U柱(C)上的色谱图Fig.9 Chromatograms of 4 types of estrogens on C18(A),C22-A(B) and C22-A/U(C) columns
3 结 论
本文建立了两亲性色谱固定相的均相合成新方法,制备了两种新型色谱固定相C22-A和C22-A/U。它们既具有C22 长链烷基末端,又具有能屏蔽载体残余活性的脲和酰胺强极性基团。通过对不同标样和实际样品的成功分离,证实两种新型色谱固定相除了疏水性和亲水性特征外,还具有较好的形状选择性,既能用于RPLC又能用于HILIC,可极大地扩大其应用范围,具有较高的实用价值和开发潜力。
猜你喜欢
陶瓷学报(2020年5期)2020-11-09 09:22:48
纺织科学与工程学报(2020年1期)2020-06-12 09:14:42
世界农药(2019年3期)2019-09-10 07:04:16
世界农药(2019年3期)2019-09-10 07:04:14
铜仁学院学报(2018年6期)2018-07-05 09:47:34
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:54
上海塑料(2015年3期)2015-02-28 14:52:05
火炸药学报(2014年3期)2014-03-20 13:17:38
无机化学学报(2014年8期)2014-02-28 17:32:48
