
线粒体质量控制系统在中枢神经系统疾病中的研究进展*
2024-01-02 06:53韩灵灵高俊玲
解剖学杂志 2023年5期
韩灵灵 高俊玲
(1 华北理工大学基础医学院,河北省慢性疾病重点实验室,唐山 063210;2 郑州大学附属南阳市中心医院急诊创伤外科,南阳 473000)
线粒体大约产生于20亿年前,属于真核细胞的核心细胞器,也是主要内膜系统之一。线粒体发挥三磷酸腺苷产生、脂质生物发生、氨基酸合成、细胞凋亡、离子稳态、抗氧化和钙清除等重要生物学作用[1]。线粒体的质量稳定和形态完整是细胞器保持数量、分布和功能正常所必需的条件。在神经元中,高度动态化的线粒体能够沿微管轨迹分裂、融合和移动,以确保它们准确分布到神经元外围。在中枢神经系统(central nervous system,CNS)中,神经元是长寿命细胞,损伤在其线粒体中可能不断累积,因此线粒体质量控制(mitochondria quality control,mitoQC)的启动和精密调控对神经元显得尤为重要。若线粒体功能障碍,神经元因极高的能量和代谢需求无法满足,导致神经元变性[2]。同时异常蛋白质聚集体和疾病相关的突变体易损害轴突线粒体运输,这进一步导致许多急性及退行性CNS疾病中线粒体功能障碍和神经元死亡。线粒体功能主要由mitoQC系统精细调节,包括线粒体不间断的生物发生、分裂、融合、自噬、有丝分裂和转运之间的动态协调循环,以维持其形态、功能完整。同时保证正常线粒体能被招募至能量需求较高的亚细胞定位,如突触末端或者轴突分支等[3],从而更好地参与线粒体内容物之间交换。事实上已证明,这其中任何节点紊乱所造成的线粒体不可逆损伤,均会诱发神经元损伤、细胞功能异常,最终导致细胞凋亡,甚至神经功能缺失[4]。
现探讨mitoQC破坏与CNS疾病之间的内在联系,以及如何维持和/或恢复神经元线粒体稳态及相关细胞生命过程的治疗策略,重点关注近年来mitoQC在几种常见CNS疾病中的研究进展。
1 线粒体质量控制体系
1.1 线粒体生物发生
线粒体生物发生(mitochondrial biogenesis,MB)是通过原有线粒体生长和分裂形成新线粒体的动态过程。MB是一个多方面的过程,涉及高度调控的转录事件、脂质膜和蛋白质合成/组装以及线粒体DNA(mitochondrial DNA,mtDNA)复制的整合。AMP活化激酶(AMPK)-PGC-1α-NRF-TFAM轴和AMPK-SIRT1-PGC-1α-NRF-TFAM轴是调节MB的2条主要通路。前者激活导致mtDNA和蛋白质合成及产生新线粒体;后者则在稳定缺陷细胞能量代谢中起关键作用[5](图1)。PGC-1α作为MB的“主调节器”,主要位于胞质,一旦PGC-1α被磷酸化后或去乙酰化激活后,则移位细胞核上调MB[6]。
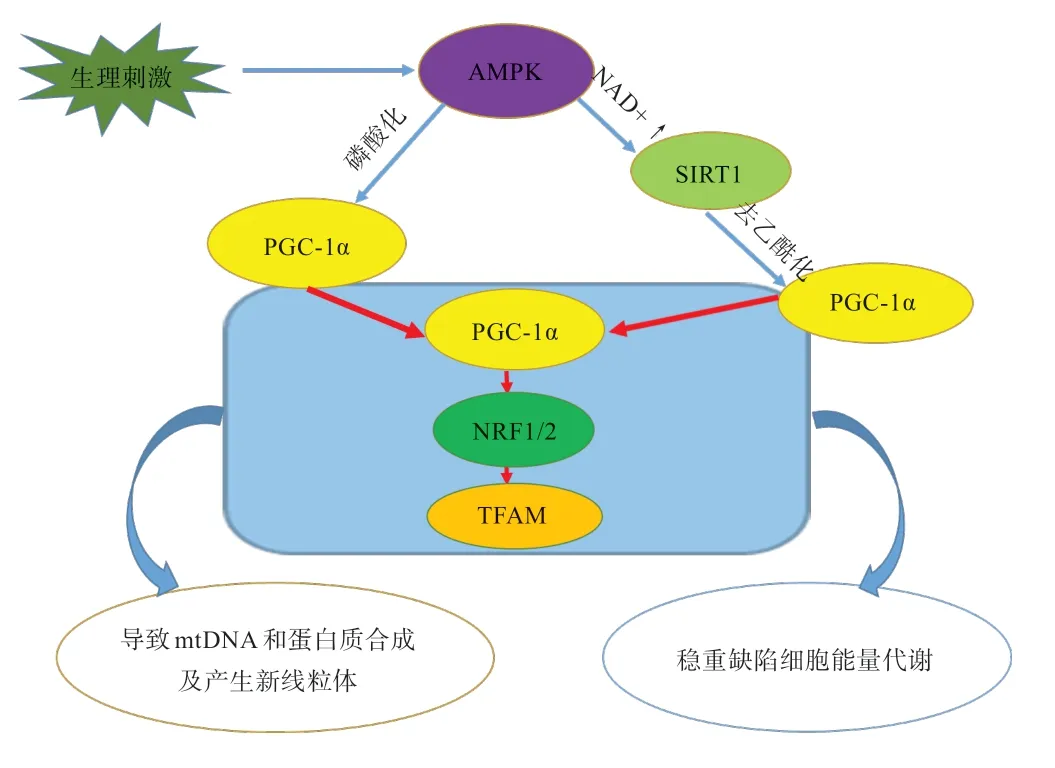
图1 调节MB的主要通路
1.2 线粒体动力学
线粒体作为真核细胞的“动力工厂”,是高度动态化的双膜细胞器,可根据宿主细胞环境需要改变其形态特征。线粒体动力学是由分裂和融合事件决定的动态循环过程。融合是指交融相毗邻去极化线粒体,混合其内容物以稳定膜电位。相反,分裂是将受损部分从整体中分离出去,并通过线粒体自噬途径进行降解,完整部分进行融合。线粒体动态特性对神经元活动至关重要,融合和裂变的不平衡可能导致线粒体分布异常和细胞功能缺陷[7]。
线粒体分裂和融合过程通过一组包含GTP酶结构域的蛋白质介导。此外,它们还共享其他域:一个中间域、一个可变域和一个GTP酶效应域。该家族的所有成员都能够通过激活可变域与膜结合[8]。主要由线粒体动力相关蛋白1(mitochondrial motility-related protein 1,Drp1)和线粒体分裂蛋白1(mitochondrial fission protein 1,Fis1)主导线粒体分裂过程,线粒体融合则由线粒体融合蛋白1/2(mitochondrial fusion protein 1/2,Mfn1/2)介导线粒体外膜融合及视神经萎缩蛋白1(optic atrophy 1,Opa1)调节线粒体内膜融合来控制[9](图2)。Drp1主要存在于胞质,募集到OMM与Fis1结合启动分裂。Drp1参与调节线粒体自噬,最终触发内源性凋亡通路。在人的Drp1的点突变足以导致致命性的大脑发育异常,分裂的维持可以调节线粒体质量和生物能量特性,分裂的失调会导致神经元疾病。系统性缺失Mfn1/2和Opa1对小鼠胚胎尤为致命,证明了它们在胚胎发生和早期发育过程中的根本重要性[10]。
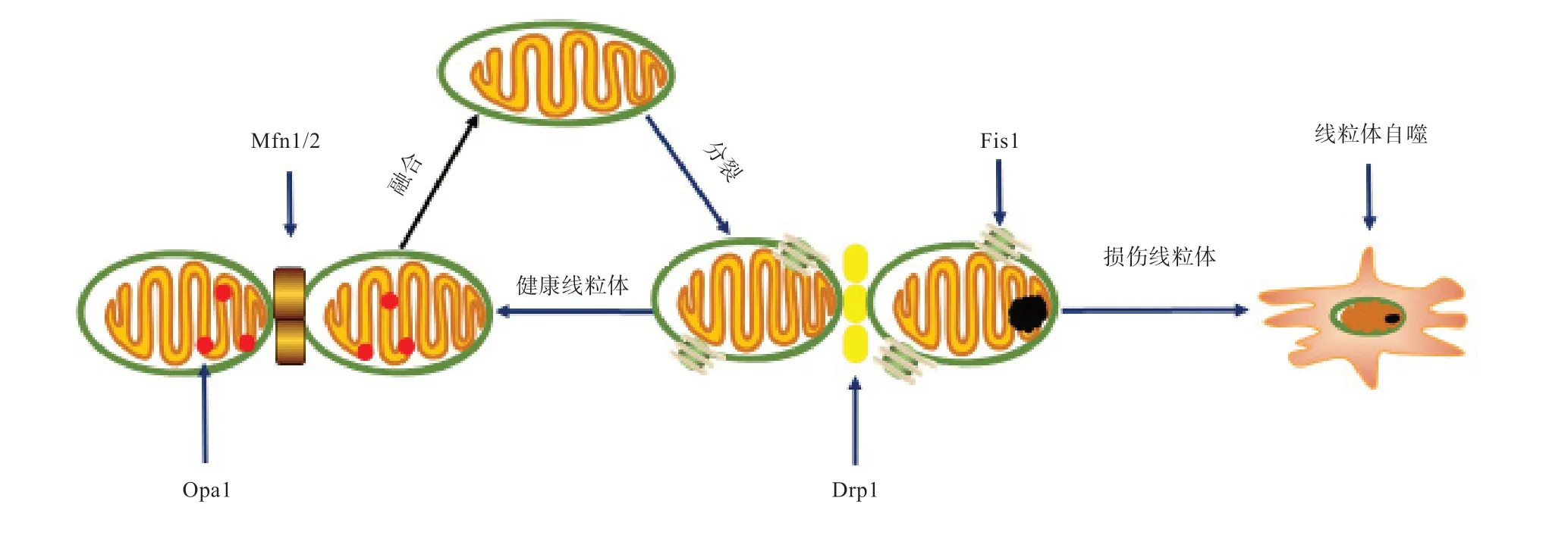
图2 线粒体动力学和自噬
1.3 线粒体自噬
自噬是一种基本生理过程,通过清除受损细胞器和异常聚集细胞溶质蛋白维持稳态。自噬涉及一系列连续事件,包括双膜形成、伸长、囊泡成熟,最终将目标物质递送至溶酶体进行降解和回收作为能源[11]。线粒体自噬过程中,线粒体被吞噬细胞吞噬形成自噬体,随后与溶酶体融合。
线粒体自噬主要通过3种机制执行诱导:第一,泛素介导线粒体自噬通路。①PINK1-Parkin通路。健康线粒体中,PINK1通过复杂加工和降解维持低水平,在线粒体外膜上保持稳定。线粒体受损后膜电位丢失,pten诱导的推定激酶1(PTEN-induced putative kinase 1,PINK1)在膜上累积,将多面泛素连接酶Parkin募集至受损线粒体并磷酸化(Ser65位点),进而刺激线粒体外膜底物蛋白泛素化启动自噬,促使自噬体形成,最终与溶酶体融合完成线粒体降解(图3)。②线粒体E3泛素连接酶1(mitochondrial E3 ubiquitin ligase 1,MUL1))与Parkin底物Drp1、Mfn1/2相互作用,可直接与神经递质γ-氨基丁酸(GABA)及其突触靶点A型GABA受体(GABAA受体)相关蛋白(GABARAP)结合,以促进自噬吞噬;GABARAP是Atg8家族的成员,在自噬和线粒体自噬中起关键作用(图3)。第二,受体介导线粒体自噬通路。受体锚定线粒体外膜内,包括NIP3样蛋白X(NIX,也称为BNIP3L)、含有1的FUN14结构域(FUNDC1)和FK506结合蛋白8(FKBP8)。NIX/BNIP3L或FUNDC1与吞噬细胞上的LC3或GABARAP的结合介导靶向功能失调的线粒体进行自噬[12](图3)。第三,脂质介导线粒体自噬。通常存在于线粒体内膜磷脂中的心磷脂外化到线粒体外膜是脂质介导的线粒体自噬的独特机制。神经元内心磷脂在线粒体损伤时由线粒体内膜外化到线粒体外膜,通过与LC3相连启动线粒体自噬,介导受损线粒体重新聚集到自噬途径中,从而起到消除信号作用[13](图3)。
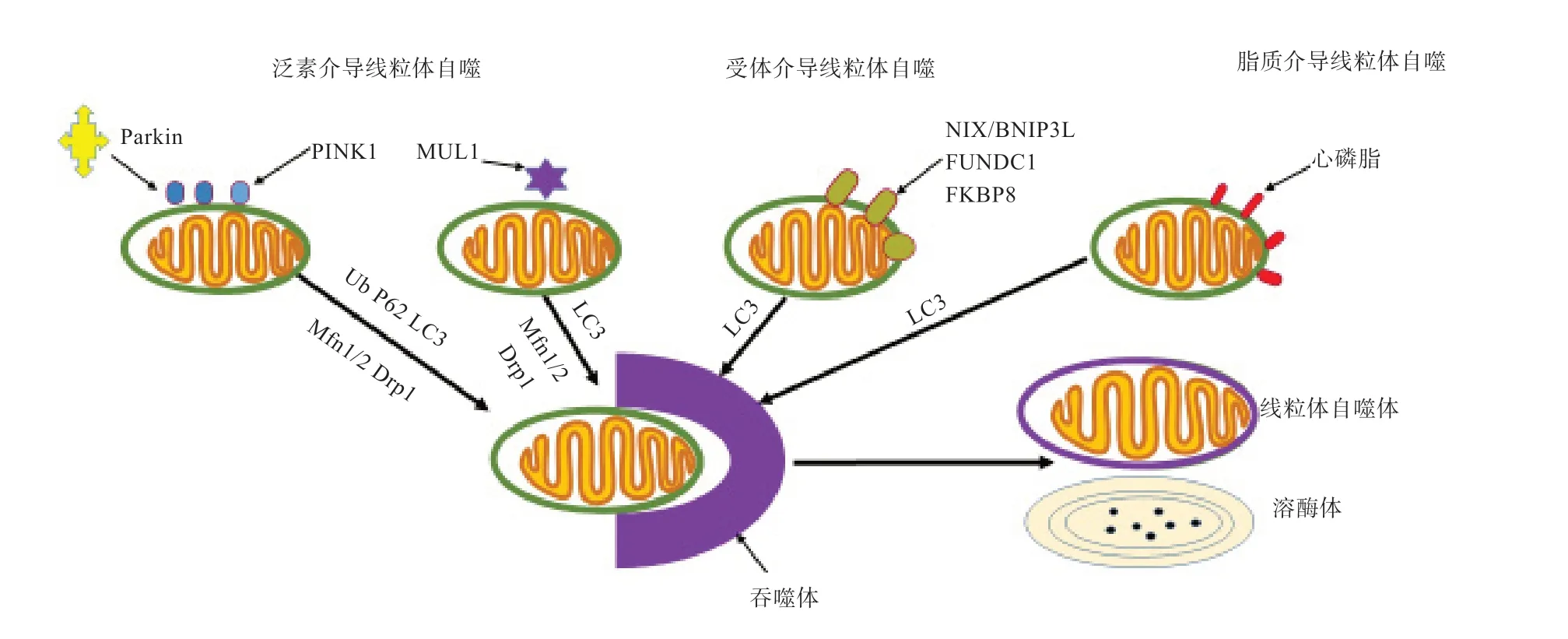
图3 线粒体自噬机制
1.4 线粒体有丝分裂
线粒体有丝分裂是一种由迁移酶体介导的线粒体质量控制过程。线粒体作为真核细胞的重要细胞器,受到严格的质量控制[14]。线粒体受损时需要被清除,以维持线粒体池的质量。在轻度线粒体应激下,受损的线粒体被运输到迁移体中,随后从迁移细胞中被丢弃。大多数已知的去除受损线粒体的机制是基于蛋白酶或蛋白酶体介导的线粒体或线粒体蛋白质的降解。线粒体可以通过各种机制从细胞中脱落[15]。线粒体的脱落可有助于线粒体质量控制[16]或作为一种机制来调节线粒体在细胞之间的水平转移[17]。
迁移体是新发现的一种由迁移细胞产生的细胞器[18]。当细胞迁移时,尾巴上会留下细长的收缩纤维。迁移体的产生与细胞的迁移高度相关。在细胞迁移过程中,回缩纤维从细胞的后端被拉出,并在回缩纤维上生长出称为迁移体的大泡状结构。当细胞移开时,回缩纤维断裂,留下迁移体。细胞内容物,如囊泡和胞浆,可以通过迁移体从细胞中释放出来。迁移体的形成依赖于迁徙。使用肌球蛋白Ⅱ抑制剂Blebbistatin或通过调节细胞黏附来阻止迁移,可以抑制迁移小体的形成[19]。最近,迁移体已被证明在斑马鱼器官发生中发挥关键作用[20]。到目前为止,关于迁移体如何发挥其生物学功能,已经出现了3种作用模式。首先,迁移体可保留在迁移细胞的迁移路径上,在细胞离开后充当位置信号。其次,迁移体充当垃圾处理机制,通过该机制将受损的细胞器从细胞中逐出。最后,迁移体可介导 RNA 和蛋白质等细胞内容物的横向或水平转移。最新研究结果表明,线粒体有丝分裂和自噬可能作为线粒体应激的两层系统。在这个系统中,有丝分裂处理轻微的线粒体应激,而线粒体自噬则处理灾难性的线粒体损伤。
现阶段,迁移体在疾病中作用的研究仍处于起步阶段。研究表明脑卒中患者大脑中存在迁移体[21]。也有研究证实受损的肾足细胞比健康的足细胞产生更多的迁移体,尿足细胞迁移体有可能用作检测早期足细胞损伤的诊断标记物[22]。此外,考虑到迁移体在细胞间通讯和维持细胞稳态中的生理功能,预计未来迁移体将与涉及迁移细胞的疾病有关,例如肿瘤转移、免疫紊乱和发育障碍。
2 线粒体质量控制与CNS疾病
CNS是由脑和脊髓组成,是人体神经系统的最主体部分。急性CNS疾病非常常见,包括脑出血(intracerebral hemorrhage,ICH)、自发性蛛网膜下腔出血(spontaneous subarachnoid hemorrhage,SAH)、创伤性脑损伤(traumatic brain injury,TBI)和缺血性脑卒中等[23]。CNS退行性疾病是指一组由慢性进行性的中枢神经组织退行性变性而产生的疾病的总称。病理上,可见脑和(或)脊髓发生神经元的退行变性和丢失。神经元死亡和线粒体功能障碍是导致急性和退行性CNS疾病患者神经功能障碍的重要因素[24,25]。线粒体质量受损,表现为膜电位丧失、ATP减少、形态异常、ROS激增、Ca2+缓冲能力受损及促凋亡分子增加,最终导致细胞死亡。因此,细胞启动mitoQC系统来克服细胞线粒体缺陷,它同时代表着生物发生和降解之间的净平衡,是维持线粒体数量、分布和功能完整的主要调控机制[5]。
2.1 mitoQC与脑卒中
线粒体是细菌来源的细胞器,被认为是细胞的能量发生器。近年来,由于线粒体已被确定在多种脑部疾病病理学中发挥核心作用,例如出血性及缺血性卒中、TBI、阿尔茨海默病(Alzheimer's disease,AD)和帕金森病(Parkinson's disease,PD)等。成年人大脑约占体质量的2%,却消耗身体所需能量的20%。线粒体提供能量占大脑总能量的主要部分,对于确保神经元的兴奋性和存活至关重要[26]。线粒体钙超载、活性氧(reactive oxygen species,ROS)过量产生、线粒体通透性转换孔开放和损伤相关分子模式释放将线粒体置于一系列复杂的偶然相互作用的中心。
线粒体功能障碍的抑制有助于脑卒中患者的恢复。线粒体自噬的恢复降低了原发性高血压和自发性卒中模型中的卒中发生概率。研究表明,Drp1选择性抑制剂Mdivi-1(mitochondrial division inhibitor 1,Mdivi-1)治疗可改善大脑缺氧/葡萄糖剥夺后的结果,并在几种脑卒中和TBI模型中减轻脑损伤[27]。此外,最新研究证明,ICH小鼠模型中Mdivi-1可逆转Drp1易位和线粒体形态学改变,并能改善突触功能障碍、血脑屏障破坏和脑水肿[28]。在体内卒中模型中研究受损线粒体动力学的恢复,通过下调Drp1表达减少线粒体分裂可使缺血性脑卒中梗死面积减小[9]。相反,Opa1过表达增强线粒体融合可减轻大鼠脑缺血后的脑水肿。这些研究表明线粒体稳态和平衡的恢复可能对脑卒中产生神经保护的结论。
2.2 mitoQC与TBI
TBI是外力作用于脑组织而导致的脑损伤,除原发性脑损伤外,局部脑组织损害、水肿和高颅压对周围脑组织形成的继发性脑损伤进一步加重病情。由于病情特殊性及病理生理反应复杂,CNS常出现不可逆性损伤,致残及病死率高。TBI死亡的主要原因是继发性脑损伤,所有这些都包含一系列有害的生化和病理生理应激源为特征的继发性事件波,称为继发性损伤。这些次级压力因素导致持续的细胞死亡和功能障碍,包括谷氨酸兴奋性毒性、线粒体功能障碍、自由基介导的氧化损伤、炎症及凋亡细胞死亡信号通路的激活[29]。
研究认为,TBI后受伤大脑的线粒体功能受到相当大的损害,例如钙超载、ROS产生增加、过氧化物酶体增殖物激活受体-γ共激活因子1α(peroxisome proliferator activated receptor-γ coactivator 1α,PGC-1α)表达降低和线粒体DNA受损[30]。在TBI动物模型中,在脑损伤后1 h,Drp1的表达开始上调,在24 h达到高峰,Mdivi-1抑制Drp1表达可显著减轻脑损伤后的行为障碍和脑水肿,减轻线粒体的形态变化,减少脑损伤后细胞死亡和损伤体积。同时,mdivi-1也抑制了TBI诱导的细胞色素c从线粒体释放到细胞质,以及半胱氨酸蛋白酶-3的激活[31]。此外也有研究表明,Mdivi-1可有效改善认知障碍和焦虑样行为。这一部分作用可能是通过挽救脑外伤后海马区的神经发生和抑制神经元凋亡来实现[27]。总而言之,笔者可以认为TBI后线粒体功能障碍的严重程度是细胞存活、组织保留和功能恢复的关键决定因素。
2.3 mitoQC与AD
神经退行性疾病是一组无法治愈的异质性疾病,其特征是CNS神经元功能和结构的进行性丧失,主要包括AD、PD、亨廷顿病等。确切发病原因不明。然而,多个病理学和生理学证据表明,线粒体质量控制体系紊乱在神经退行性疾病的衰老和发病机制中起着至关重要的作用。
AD包括家族性AD和散发性AD,是一种与年龄相关的神经退行性疾病,其特征是进行性认知障碍、记忆力减退和行动不便。AD的发病机制复杂,涉及淀粉样蛋白β(Aβ)代谢异常,导致Aβ斑块沉积、Tau蛋白过度磷酸化、神经原纤维缠结沉积、反应性胶质细胞改变等病理现象。多项研究报告AD患者死后大脑中Aβ斑块和磷酸化Tau聚集在突触间隙、高代谢区域,从而阻碍突触线粒体和神经传递。这种障碍导致AD患者的神经元功能障碍和认知能力下降,Aβ聚集体还促进ROS的产生,进一步破坏mtDNA,同时也促进额外的Aβ形成[32]。
线粒体动力学的不平衡是AD的主要发病机制之一,即线粒体分裂的上调、线粒体融合的下调以及线粒体自噬的减少。细胞培养和许多AD动物模型及AD患者死后大脑中都观察到线粒体碎片和异常的线粒体分布和神经元运输[33]。AD患者大脑中,Drp1在Ser616处的磷酸化和Drp1的S-亚硝基化均显著高于对照组,证实了过度线粒体分裂的诱导。给予Mdivi-1治疗,有助于恢复β-淀粉样蛋白前体蛋白(βAPP)的蛋白水解衍生物淀粉样蛋白-β肽(Aβ)介导的线粒体功能障碍和神经元突触抑制[34]。此外,AD中的异常线粒体积累被认为是由多种线粒体自噬缺陷机制引起,在线粒体自噬通路中,PINK1和PD蛋白2的突变导致明显的线粒体功能障碍,从而导致肌肉和神经元的退化[35]。因此,这些研究表明,缺陷的线粒体自噬可能是AD大脑中的早期事件,并且线粒体功能障碍与AD发病机制密切相关[36]。
2.4 mitoQC与PD
PD是全球最常见的与年龄相关的运动恶化神经退行性疾病,影响超过400万人。PD特征是黑质多巴胺能(dopaminergic,DA)神经元首先退化,当大约70%的神经元丢失时,大脑和肌肉细胞之间联系减弱,导致典型运动症状:如静止性震颤、僵硬、运动迟缓和姿势不稳。PD病因有线粒体功能障碍、氧化应激、慢性炎症、异常蛋白质折叠和异常蛋白质聚集[37]。而线粒体功能障碍越来越被认为是PD神经元易感性的关键危险因素。
对PD研究显示,PD与线粒体直接相关的第一个关键结果是线粒体自噬相关蛋白Pink1和Parkin的发现,这表明线粒体自噬在PD中尤为重要。在PD中,PARK6(编码PINK1)和PARK2(编码Parkin)基因突变会导致5%的家族性PD,而PINK1突变则与常染色体隐性遗传性少年型PD有关。已有研究表明Pink1与线粒体中的α-突触核蛋白本身相互作用,并通过刺激线粒体自噬清除,从而降低由α-突触核蛋白引起的毒性[38]。除Pink1和Parkin外,PD患者基因突变至少包括SNCA、GBA、DJI、LRRK2。这些基因大多数相关联或定位于线粒体,而PD中线粒体异常状态,包括线粒体复合物I活性降低、膜电位下降、活性氧大量生成、线粒体体动力学改变及DNA突变等,也反向证明了这种联系的存在。
3 总结与展望
CNS中的细胞严重依赖线粒体的完整性,线粒体功能障碍的许多方面都表现在CNS疾病中。神经退行性疾病,包括 AD、PD,其特征都是特定大脑区域进行性神经元变性。迄今为止,治疗策略对这些神经退行性疾病无效。线粒体调节细胞代谢,协调细胞死亡,在激活途径中发挥作用,并且对于维持神经元完整性至关重要。在过去10年中,越来越多的证据表明,线粒体动力学和线粒体自噬是解释这些疾病中线粒体和神经元功能障碍的共同和初始特征。值得注意的是,线粒体动力学和线粒体自噬的操纵已被证明对这些疾病具有保护作用,这为开发有效的治疗方法提供了新的方向。
此外,近年来,迁移体作为一种独特的细胞外囊泡开启了新的视野。尽管其特性和功能已逐渐阐明,但迁移体的生物发生机制,特别是病理生理学方面仍处于起步阶段,许多重要问题仍未得到解答。迁移体很可能在涉及迁移细胞的其他生物学过程中发挥重要作用,包括神经元变性、免疫反应、血管生成、组织再生和肿瘤转移等。建立各种动物模型来研究迁移体在这些生物过程中的作用将是至关重要的。最近的研究描述了迁移体在疾病中的可能作用,并讨论了迁移体的诊断和治疗潜力。在考虑可能的诊断和治疗潜力之前,还需要更多确凿的证据。只有更好地了解迁移体在各种生理和病理过程中的作用,才能产生基于迁移体的诊断工具和治疗方法。
猜你喜欢
自然杂志(2021年6期)2021-12-23
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
中国生殖健康(2019年12期)2019-01-07
现代装饰(2018年5期)2018-05-26
分子影像学杂志(2015年3期)2015-12-04
电源技术(2015年5期)2015-08-22
西南军医(2015年3期)2015-04-23
弹箭与制导学报(2015年1期)2015-03-11
癌变·畸变·突变(2014年1期)2014-03-01
