
棉花衣分和铃重的全基因组关联分析
2024-01-01 00:00:00杜霄龙遗磊谭彦平李丽丽王胤金珅杨伊楠艾先涛
棉花学报 2024年6期
关键词:衣分







摘要:【目的】对棉花衣分和铃重进行全基因组关联分析,挖掘相关的候选基因,为通过分子标记辅助选择和分子设计育种提高棉花产量提供遗传基础。【方法】利用300份陆地棉种质资源重测序(10×)数据和3 055 642个高质量单核苷酸多态性(single nucleotide polymorphism, SNP)对2年5个环境及最佳线性无偏预测值(best linear unbiased predictive value, BLUP)的衣分和铃重进行了全基因组关联分析,检测相关的显著关联位点和候选基因。【结果】衣分和铃重在不同环境下存在较广泛的变异,衣分平均变异系数为9.40%,遗传力为92.81%;铃重平均变异系数为11.96%,遗传力为86.67%。不同环境间,群体的铃重呈极显著正相关关系,衣分也呈极显著正相关关系。群体结构分析、主成分分析和系统发育分析将300份陆地棉分为6个亚群,全基因组关联分析共检测到223个数量性状位点(quantitative trait locus, QTL)与衣分相关,89个QTL与铃重相关。对衣分中筛选到的3个稳定的QTL qLP_Gh5.18、qLP_Gh12.43和qLP_Gh17.2进一步分析,发现17个相关候选基因;对铃重中筛选到的2个稳定的QTL qBW_Gh7.5和qBW_Gh19.5进一步分析,发现8个相关候选基因。【结论】在300份陆地棉群体中鉴定到5个稳定的QTL与棉花衣分和铃重关联,挖掘到25个与衣分和铃重相关的候选基因。
关键词:陆地棉;衣分;铃重;全基因组关联分析
Genome-wide association study of cotton lint percentage and boll weight
Du Xiao1, Long Yilei1, Tan Yanping1, Li Lili2, Wang Yin1, Jin Shen1, Yang Yinan1, Ai Xiantao3*
(1." College of Life Science and Technology, Xinjiang University/Xinjiang Key Laboratory of Biological Resources and Genetic Engineering, Urumqi 830000, China; 2. Kuqa Modern Agricultural Science and Innovation Center, Kuqa, Xinjiang 842000, China; 3. College of Intelligent Agriculture (Research Institute), Xinjiang University, Urumqi 830000, China)
Abstract: [Objective] This study aims to perform genome-wide asscciation study of cotton yield traits, such as lint percentage (LP) and boll weight(BW), and to mine the candidate genes, and may be helpful for improving cotton yield through molecular marker-assisted selection and molecular design breeding. [Methods] Genome-wide association study was performed using 300 upland cotton germplasms resequencing (10×) data and 3 055 642 high-quality single nucleotide polymorphism (SNP) for LP and BW in five environments and best linear unbiased predictive value (BLUP) for two years to detect significant association loci and candidate genes. [Results] The cotton LP and BW showed wide variations in different environments, with an average coefficient of variation of 9.40% and heritability of 92.81% for LP, and an average coefficient of variation of 11.96% and heritability of 86.67% for BW. BW is significantly positively correlated in different environments. It’s the same with LP. Population structure analysis, principal component analysis, and phylogenetic analysis classified the 300 upland cotton lines into six subgroups. Genome-wide association study detected a total of 223 quantitative trait locus (QTL) associated with LP and 89 QTL associated with BW. The three stable QTL qLP_Gh5.18, qLP_Gh12.43, and qLP_Gh17.2 screened in LP were further analyzed, and 17 related candidate genes were found. Two stable QTL qBW_Gh7.5 and qBW_Gh19.5 related to BW were further analyzed, and 8 related candidate genes were identified. [Conclusion] Five stable QTL were identified in 300 up land cotton lines associated with cotton LP and BW, and a total of 25 candidate genes related to LP and BW were mined.
Keywords: Gossypium hirsutum L.; lint percentage; boll weight; genome-wide association study
棉花(Gossypium)是世界上重要的经济作物和油料作物,为纺织品生产提供了天然纤维[1]。陆地棉具有高产、适应性强、分布广的特点,是栽培最广泛的棉种。随着全球人口的增长和工业化的不断发展,纺织工业对棉花的需求持续增长。培育高产的棉花品种是棉花育种者不懈追求的目标。由于长期人工选择育种,陆地棉品种遗传多样性降低、遗传背景狭窄[2],这无疑增加了通过常规育种手段提高棉花产量的难度。因此,通过全基因组关联分析(genome-wide association study, GWAS)挖掘与产量性状相关的优良基因或数量性状位点(quantitative trait locus, QTL),对于加速棉花高产分子育种具有十分重要的意义。
铃重(boll weight, BW)和衣分(lint percentage, LP)是重要的棉花产量性状,也是典型的数量性状,容易受到环境因素的影响[3]。大量学者通过分子标记技术和双亲连锁作图技术,鉴定了许多与棉花产量性状相关的QTL[4-8],这些研究为解析棉花产量性状的分子机理及分子标记辅助选择(molecular marker-assisted selection, MAS)育种提供了基础。然而,这些来自种间群体的QTL大多定位于非常大的遗传区域,往往不稳定,因此不能直接应用于陆地棉改良[9]。GWAS是以连锁不平衡(linkage disequilibrium, LD)为基础,通过检测成千上万的分子标记,筛选出与目标性状表型变异相关联的分子标记,进而分析关联分子标记对表型的遗传效应[10]。因分辨率高、成本低、不需要系谱等优势,GWAS已逐渐成为解析复杂数量性状遗传基础的有效方法,利用该方法在棉花株型[11-12]、纤维品质[13-16]、抗性[17-18]等性状的相关研究中挖掘了大量的QTL/基因。在棉花产量性状的研究中,Zhu等[19]对13个不同地点的242个棉花种质进行GWAS分析,报道了95个产量相关的非冗余QTL,其中包括12个在6个及以上环境检测到的稳定的QTL和1个重要基因Gh_A07G1389,该基因编码四肽重复样超家族蛋白,与超短纤维突变体(Liy)相关基因同源。Sun等[20]利用SNP63K阵列对719份棉花进行了基因分型,鉴定出2个提高皮棉产量的基因Gh_
D03G1064和Gh_D12G2354。Song等[21]利用SNP63K阵列对276份棉花进行基因分型,发现Gh_D05G0313和Gh_D05G1124在胚珠和纤维发育阶段高表达。Wang等[22]利用SNP80K阵列对189个棉花进行基因分型,鉴定了与BW相关的候选基因Gh_A02G1473、Gh_A10G1765和Gh_
A02G1442。这些重要候选基因的发掘为棉花高产育种奠定了坚实的理论基础。
单核苷酸多态性(single nucleotide polymorphism, SNP)的标记密度是影响GWAS准确性的主要因素之一[23],上述棉花产量性状相关研究中由于受SNP标记密度的影响,导致挖掘的候选基因数量受限。本研究以300份不同来源的陆地棉重测序(10×)数据,对5个环境下的BW和LP进行全基因关联分析,以确定遗传位点与产量性状的关系,研究结果可为棉花产量的分子育种提供理论依据。
1 材料与方法
1.1 材料
300份不同来源陆地棉种质资源材料由中国农业科学院棉花种质资源库提供(表1),其中,国内种质274份,国外种质26份。
1.2 试验设计
300份棉花材料于2022年4月10日种植在新疆库车市乌恰镇现代农业科创中心1号和2号试验田,分别用E1、E2表示这2个环境;2023年4月9日种植在新疆库车市乌恰镇现代农业科创中心1号、2号和3号试验田,用E3、E4、E5表示这3个环境。2号试验田是2022年新开垦改良的试验田,3号试验田是2023年新开垦改良的试验田,因3块试验田棉花长势有明显差异,可以视作不同的环境。每个试验田设3个重复,采用随机区组设计,行距配置为(66+10)cm,株距为10 cm,1膜6行,幅宽2.28 m,小区行长4 m。机械铺膜打孔,人工膜上点播,膜下滴灌栽培,田间管理措施同常规大田生产一致。
1.3 表型测定方法
参照杜雄明等[24]编著的《棉花种质资源描述规范和数据标准》,在吐絮期,每个材料挑选10个连续单株,分别采收每株中部果枝吐絮畅的棉铃1个,共采收10个棉铃,随后进行室内考种,考种项目包括BW和LP。
1.4 表型数据分析
采用SPSS Statistics 25和Prism 9进行相关数据的统计分析;采用软件R 4.4.0对不同环境的表型数据进行相关分析,用lme4包对各环境各性状表型的最佳线性无偏预测(best linear unbiased prediction, BLUP)值进行计算,并计算广义遗传力(H2),公式如下:
H 2=Vg/(Vg+++)
式中,Vg、Vge、Vgy、Ve、L、Y分别代表遗传方差、基因型与环境间的交互方差、基因型与年份的交互方差、环境方差、环境数和年份数。
1.5 DNA提取和基因组重测序
对每个棉花种质的单株幼叶进行取样,使用康为世纪的CWE9600 Magbead Blood DNA Kit试剂盒运用磁珠法进行DNA的提取;并通过随机DNA片段化(300~350 bp)、末端修复、加PolyA并连接测序接头,筛选300~350 bp的DNA片段,通过聚合酶链式反应扩增和纯化,从而获得测序文库。随后使用DNBSEQ-T7测序仪,采用双末端(pair-end, PE)150 bp的测序方法上机测序。
1.6 测序读长质量过滤和比对
使用fastp对原始数据进行一系列的质量控制,数据质控的标准包括:去除带接头的读长;去除未知/不确定碱基含量超过1%的读长;去除低质量(Q≤5)碱基数超过50%的读长。使用BWA" 0.7.17(MEM算法)软件将干净读长比对到陆地棉TM-1参考基因组(http://ibi.zju.edu.cn/cotton/source/TM-1_V2.1),使用GATK 4.1.8.0软件自带的模块对Bam文件进行去重,然后基于Bam文件统计各样品的测序深度、基因组覆盖度等信息。
1.7 群体SNP检测
使用软件GATK 4.1.8.0 callSNP命令调取SNP,采用VariantFiltration模块对SNP进行严格质控过滤,选择标准:群体内个体缺失率≤1 %、SNP缺失率≤1 %、次要等位基因频率(minor allele frequency, MAF)>0.05,最终保留3 055 642个高质量 SNP用于主成分分析(principal component analysis, PCA)、系统发育树、群体结构分析和GWAS分析。
1.8 群体结构和LD分析
为了从全基因组的角度阐明系统发育关系,利用Tassel软件中的邻接法(neighbor-joining method)构建系统发育树。使用软件Admixture评估群体遗传结构,假设K取值2~10,每次运行迭代10 000次。利用GCTA软件进行PCA分析以评估群体的遗传结构。使用软件PopLDdecay计算成对高质量SNP之间的LD系数(r2),结果用于估计LD衰减。
1.9 GWAS
利用3 055 642个高质量SNP对2个性状进行GWAS分析,利用全基因组高效混合模型关联软件包GEMMA 0.94.1[25](http://www.xzlab.org/
software.html),用以群体结构(Q)矩阵为协变量的一般线性模型GLM (Q)、以主成分(P)矩阵为协变量的一般线性模型GLM (P)、以群体结构(Q)和亲缘关系(K)矩阵为协变量的混合线性模型MLM (Q + K)、以主成分(P)和亲缘关系(K)矩阵为协变量的混合线性模型MLM (P + K)等4个模型进行关联分析。根据-lg(1/N)计算阈值,N为有效SNP数。本研究中使用“plink--indep--pairwise 50 10 0.1”质控后获得182 147个有效SNP,经计算阈值为5.26。
1.10 单倍型分析及候选基因的预测
GWAS分析后,对QTL进行命名,对所有环境及模型下检测到的显著SNP按照其位置进行排序,若相邻2个SNP的距离大于LD衰减距离454.6 kb,则这2个SNP属于不同的QTL,反之则视为是同1个QTL。对多环境重复检测到的稳定QTL,选择每个QTL中表型变异解释率(phenotypic variation explained, PVE)最大的SNP做单倍型分析,利用曼-惠特尼检验对不同单倍型之间的差异显著性进行检验。将棉花候选基因比对到拟南芥基因组(www.arabidopsis.org)同源基因并进行功能注释。
2 结果与分析
2.1 表型分析
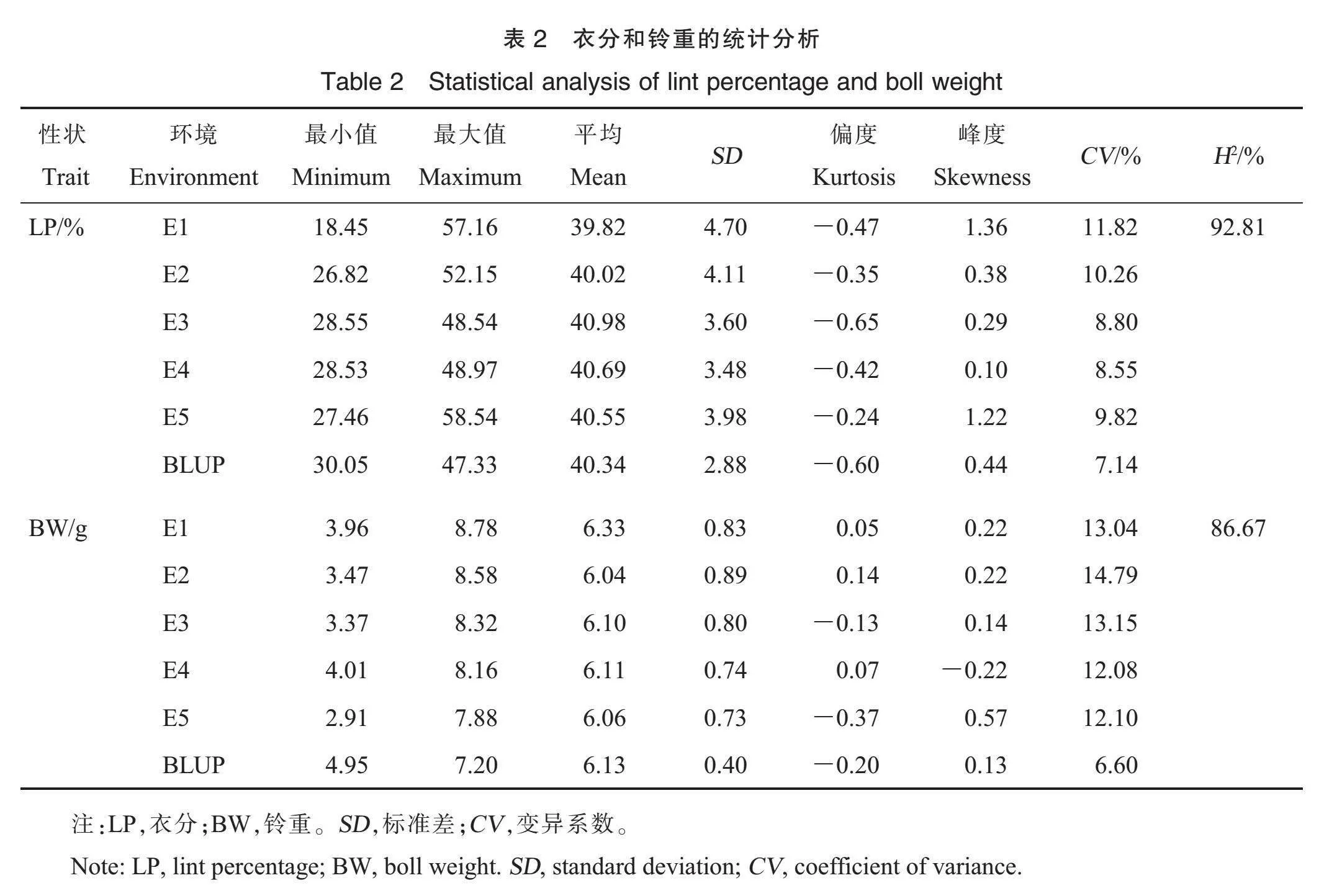
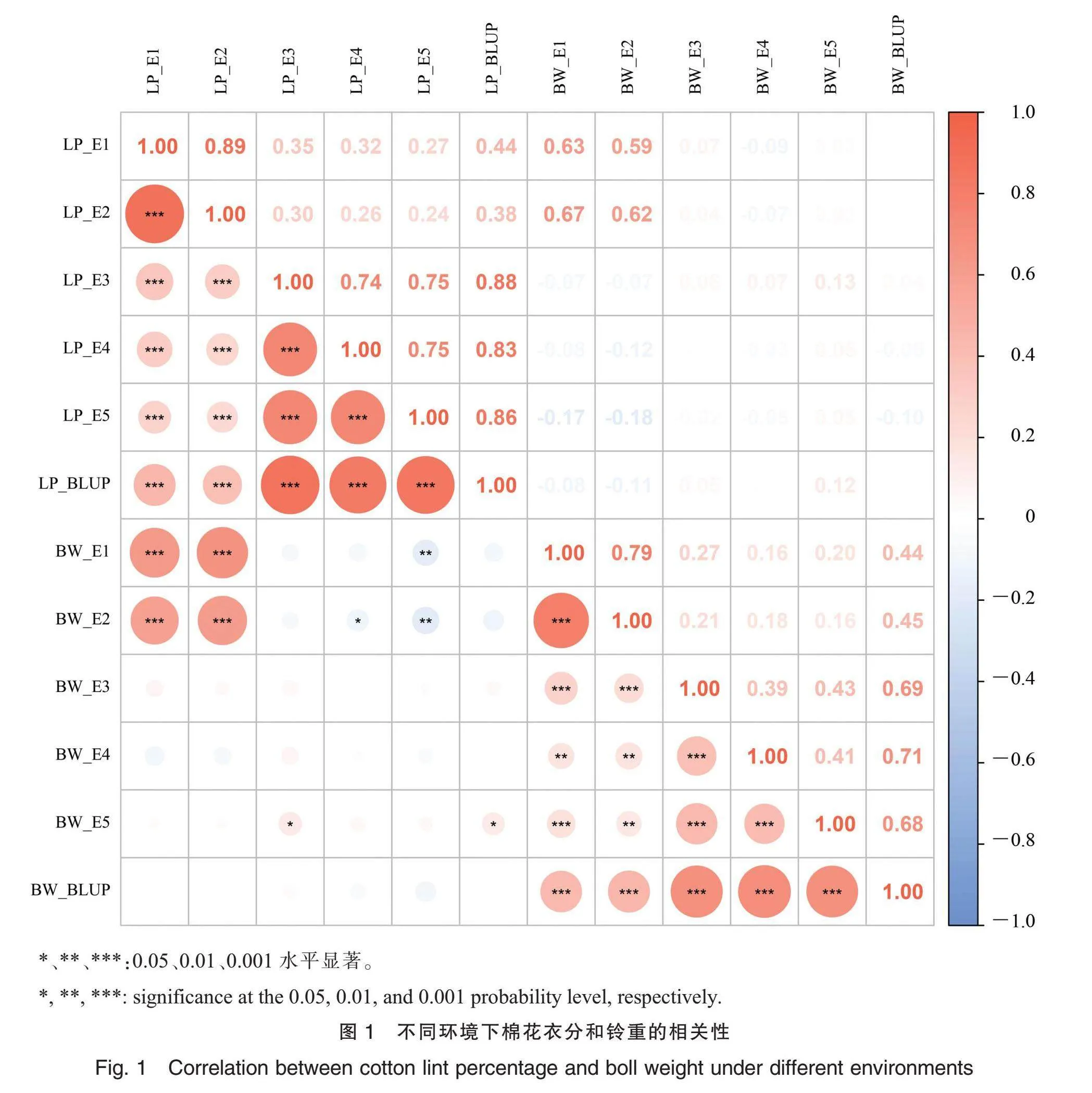
为了评估关联群体中产量性状的表型变异,对LP和BW在2年5个环境的表型值及BLUP值进行统计分析(表2)。结果表明,LP变化范围为18.45%~58.54%;BW变化范围为2.91~8.78 g。2个性状在5个环境及BLUP值下均具有较广泛的变异,LP的平均变异系数为9.40%,BW的平均变异系数为11.96%;LP的遗传力为92.81%,BW的遗传力为86.67%,表明这2个性状主要受遗传因素影响。LP的偏度为-0.65~
-0.24、峰度为0.10~1.36,BW的偏度为-0.37~
0.14、峰度在-0.22~0.57之间,两者均呈近似正态分布,符合数量性状特征。LP在5个环境下的表型值和BLUP值间均呈极显著的正相关关系,BW类似,但是LP与BW在不同环境间呈现不同的相关关系,有呈正相关关系的,如LP_E1与BW_E1、BW_E2;也有呈负相关关系的,如LP_
E5与BW_E1、BW_E2(图1)。
2.2 群体结构和LD分析
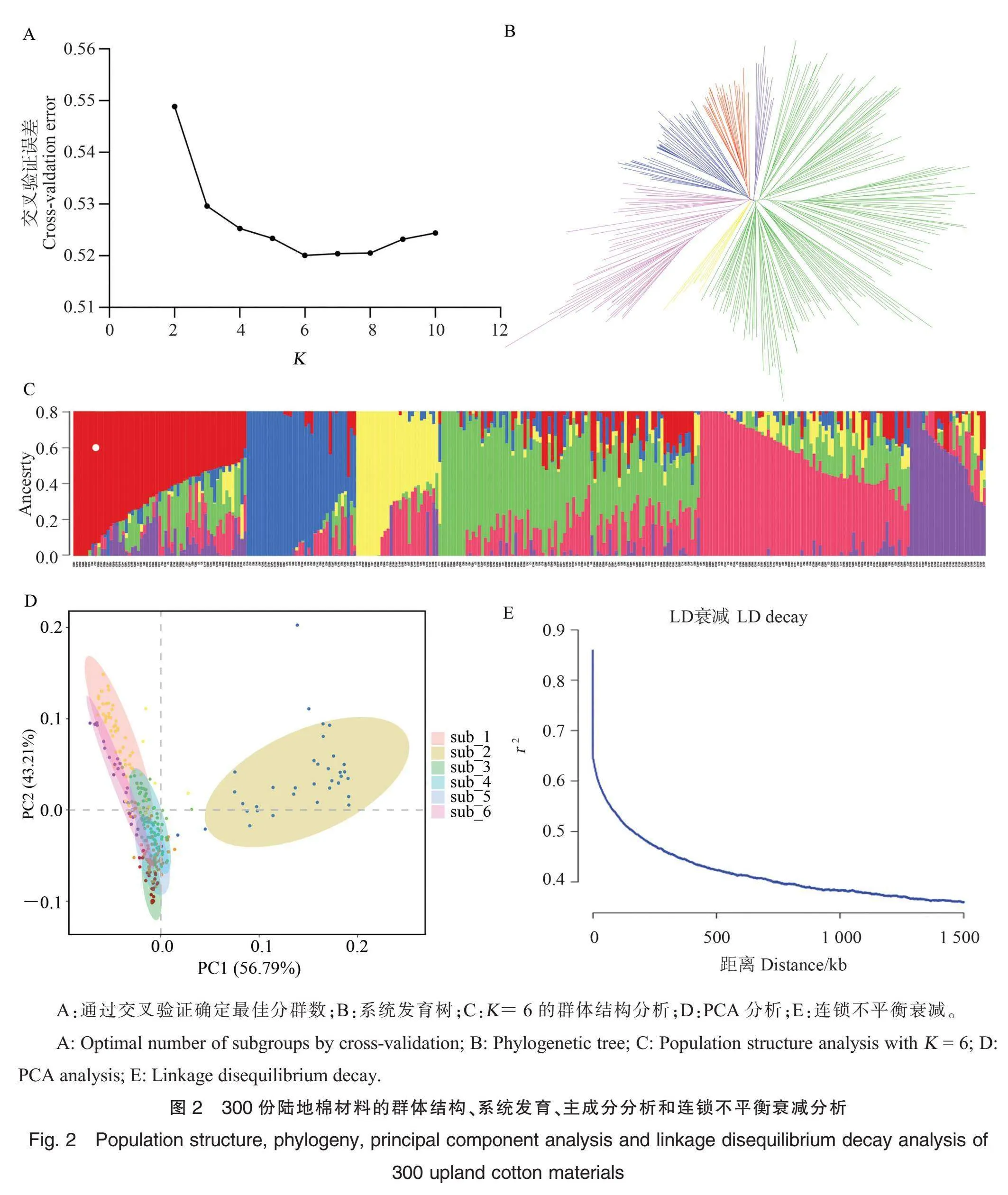
群体结构分析对后续研究不同亚群遗传信息尤为重要,可以初步推断亚群分化程度和亲缘关系,反映群体内遗传多样性。利用Admixture对群体结构进行分析,当K为6时,交叉验证误差最小,从而确定K最佳取值为6(图2A),即300份陆地棉材料分为6个亚群(图2C)。系统发育分析(图2B)和PCA分析(图2D)也将这些材料划分为6个亚群。根据PopLDdecay计算r2最大值为0.86,衰减一半时对应的距离为454.6 kb,即300份陆地棉材料的LD衰减距离为454.6 kb(图2E),LD衰减距离中等,适合进行后续的GWAS。
2.3 全基因组关联分析
利用3 055 642个高质量的SNP对2年5个环境的LP、BW表型数据及其BLUP值,使用GLM (Q)、MLM (Q+K)、GLM (P)和MLM (P+K) 4种模型进行GWAS分析。GLM (Q)模型检测到8 358个与LP显著关联的SNP,MLM (Q+K)模型检测到198个,GLM(P)模型检测到17 355个,MLM (P+K)模型检测到2 385个,共检测到28 296个SNP与LP显著关联(包含不同模型重复检测的SNP)。对于BW,GLM (Q)模型检测到680个显著关联SNP,MLM (Q+K)模型检测到34个,GLM (P)模型检测到754个,MLM (P+K)模型检测到36个,共检测到1 504个SNP与BW显著关联。按照454.6 kb的LD衰减距离(图2E),将与LP显著关联的SNP整合到223个QTL中,解释了6.67%~14.35%的PVE,将与BW显著关联的1 504个SNP整合在89个QTL中,解释了6.66%~10.24%的PVE。其中,被重复检测的QTL共91个,88个与LP关联、3个与BW关联。图3展示了这91个QTL在26条染色体上的分布情况,与LP关联的QTL在除A01外的染色体均有分布,其中A02染色体上数量最多,为23个;A12次之,共7个;A03、A04、A09、A10、A13、D01和D09号染色体上各有1个QTL。A07、D06和D11号染色体上各有1个与BW关联的稳定QTL。
2.4 候选基因的预测
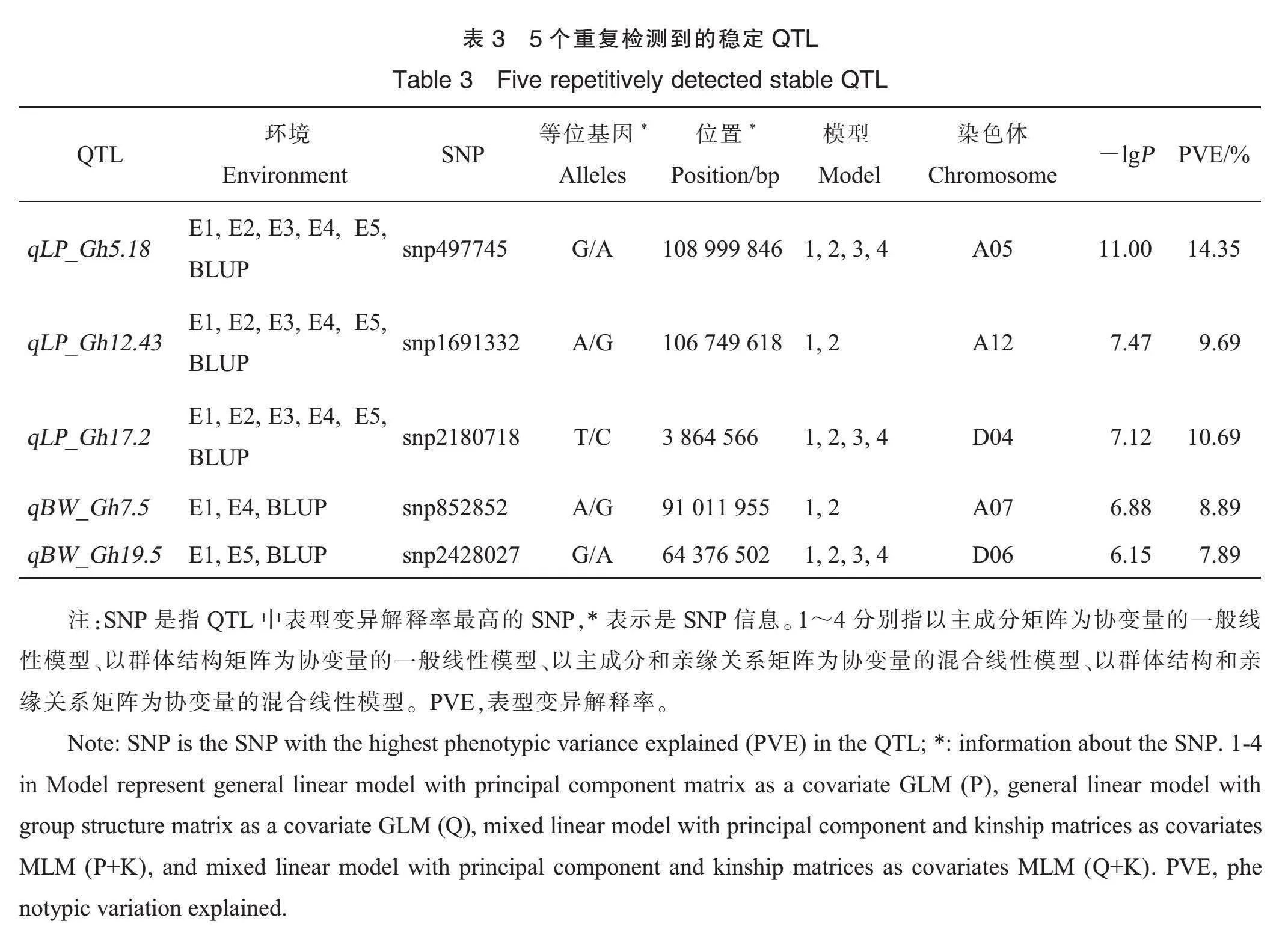
为了筛选与LP和BW相关的稳定QTL,从上述91个QTL筛选在5个环境以及BLUP值中重复检测到3次及以上的稳定QTL(表3),获得3个与衣分相关的QTL:qLP_Gh5.18、qLP_Gh12.43、qLP_Gh17.2,2个与BW相关的QTL,分别是qBW_Gh7.5和qBW_Gh19.5。
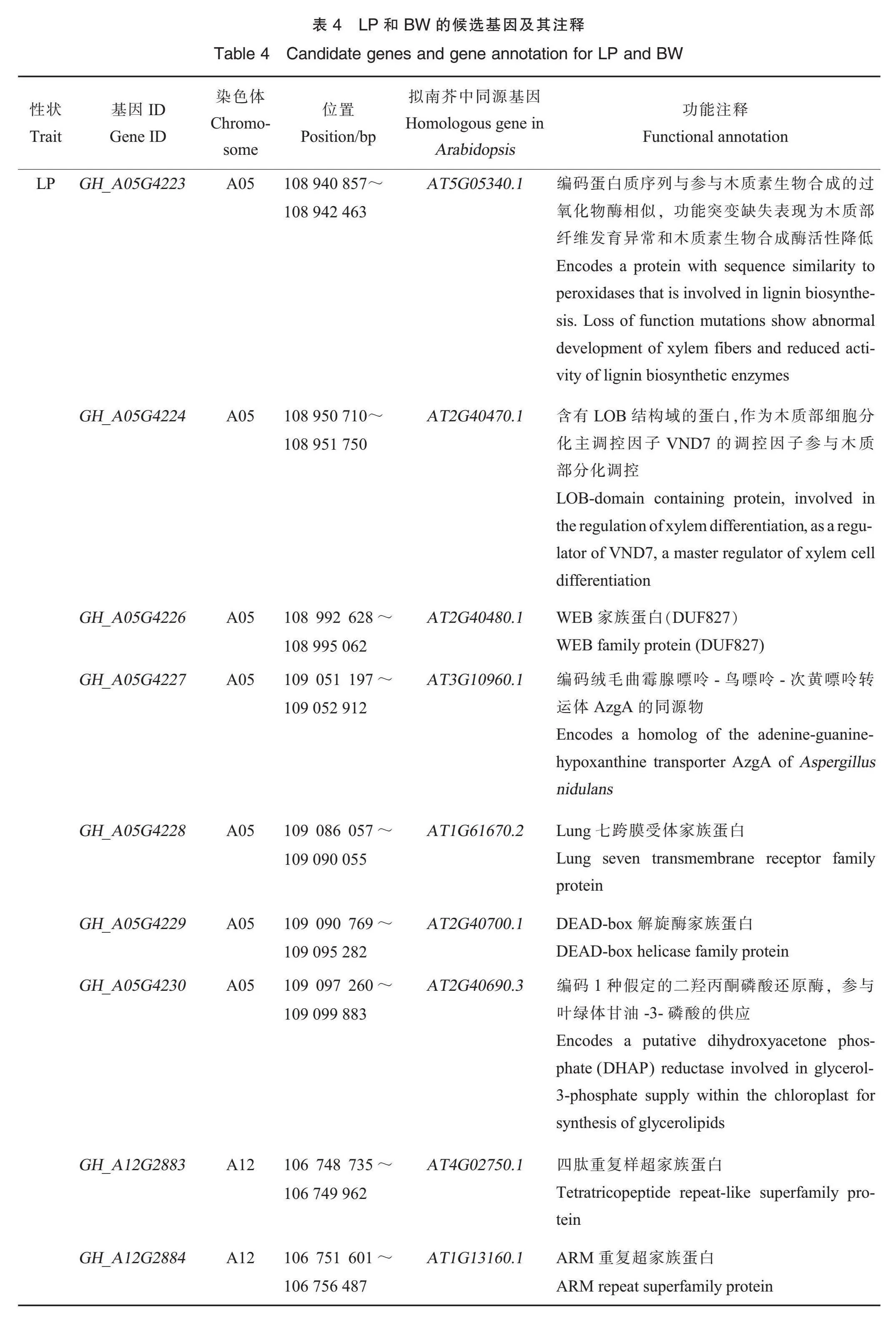
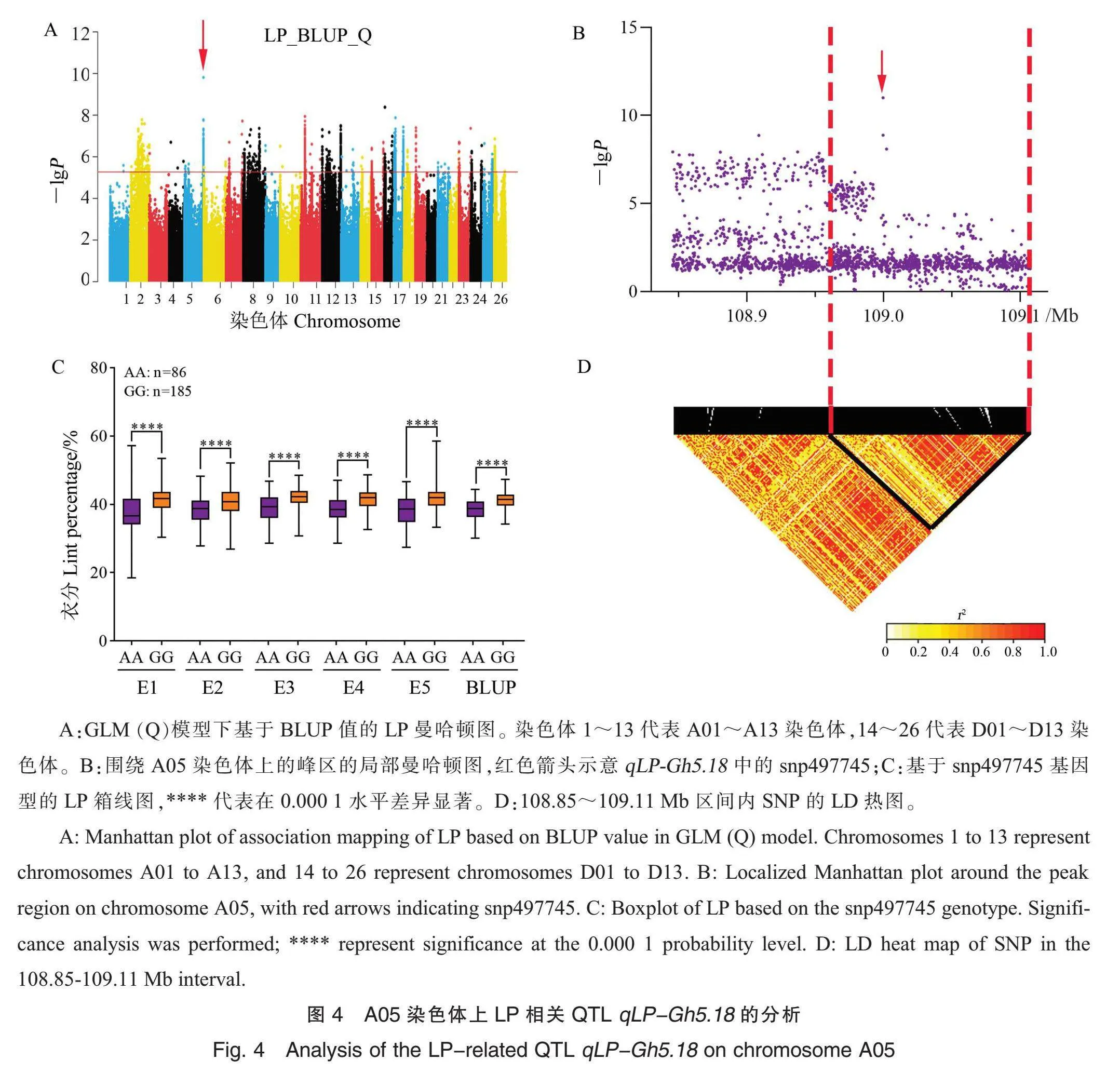
对A05染色体上qLP-Gh5.18进行分析,该QTL在5个环境及BLUP值下在4种模型中均被检测到(图4A和表3)。其中,snp497745解释14.35%的PVE,是qLP_Gh5.18中PVE最高的SNP位点,对该SNP进行后续分析(后文也是用QTL区间内PVE最高的SNP进行分析)。局部曼哈顿图(图4B)表明在A05染色体上108.85~109.11 Mb区间出现明显峰值,对此区间内的LD程度进一步分析并结合LD热图,将候选区间缩小为160 kb(图4D)。在此区间内共注释了7个候选基因(表4),结合拟南芥注释信息,推测GH_A05G4223与拟南芥基因AT5G-
05340.1同源,拟南芥AT5G05340.1编码产物与参与木质素生物合成的过氧化物酶相似,其功能突变缺失导致木质部纤维发育异常以及木质素生物合成酶活性降低。GH_A05G4224编码含有侧器官边界(lateral organ boundaries,LOB)结构域的蛋白,作为木质部细胞分化主调控因子VND7的调控因子,参与木质部分化调控。GH_A05G4226编码WEB家族蛋白DUF827;GH_A05G4227编码绒毛曲霉腺嘌呤-鸟嘌呤-次黄嘌呤转运体AzgA的同源物;GH_A05G4228在拟南芥中的同源基因编码Lung七跨膜受体家族蛋白;GH_A05G4229编码DEAD-box解旋酶家族蛋白;GH_A05G4230编码1种假定的二羟丙酮磷酸还原酶,参与叶绿体内甘油-3-磷酸的供应。单倍型分析发现携带AA单倍型的棉花品种的LP极显著低于携带GG单倍型的品种(图4C)。
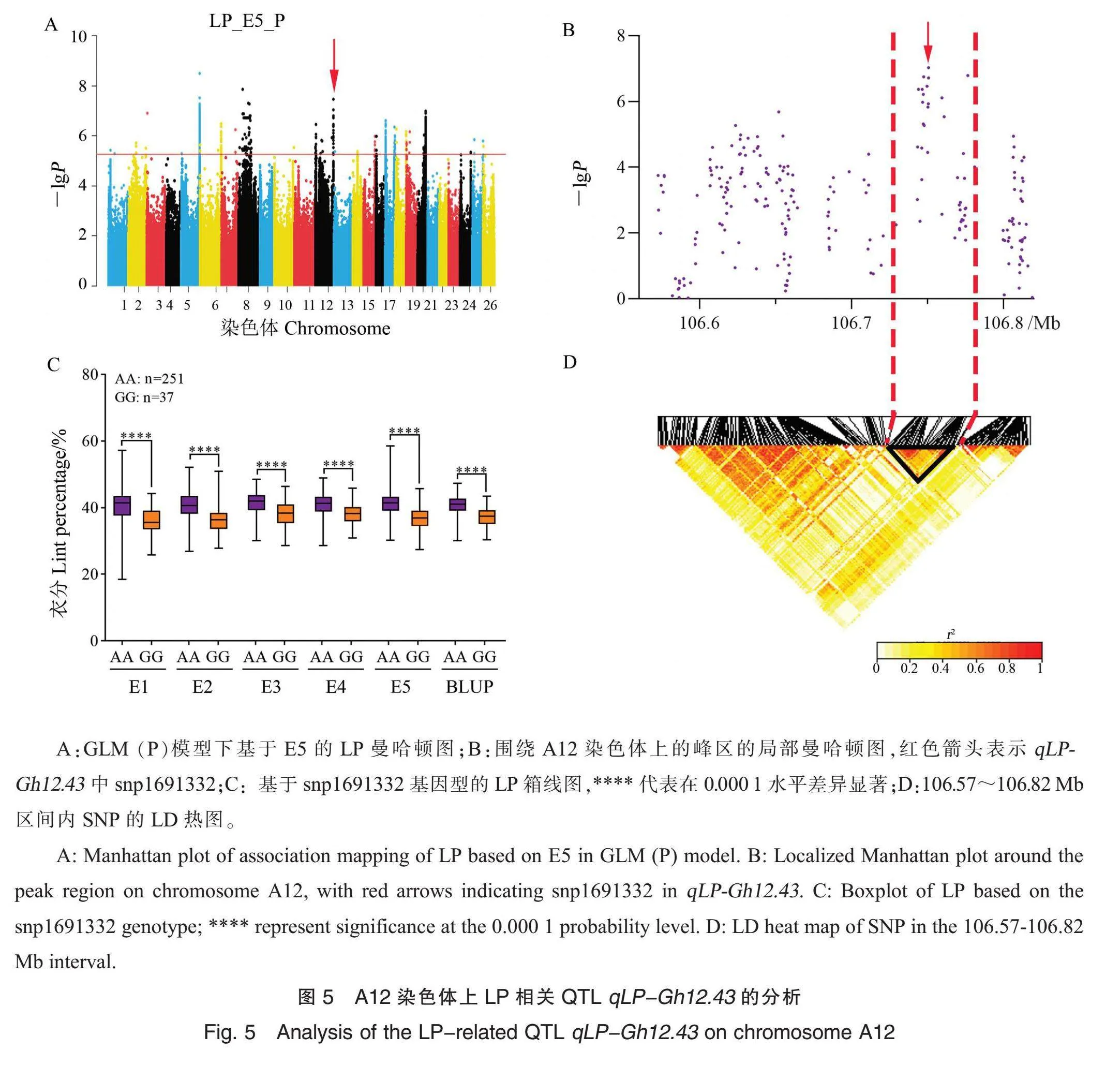
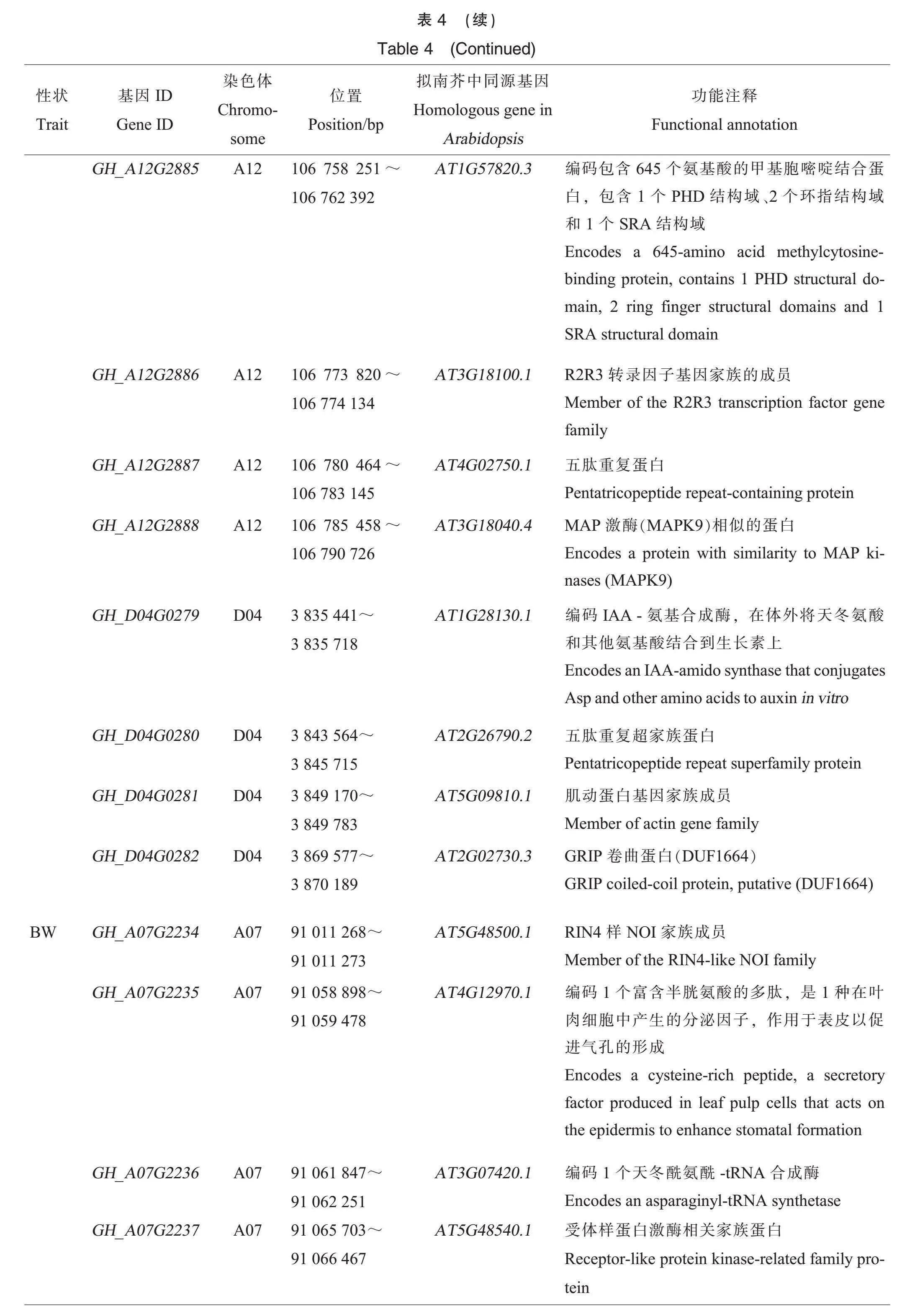
对A12号染色体上qLP-Gh12.43进行分析,该QTL在GLM (P)和GLM (Q)模型下在5个环境及BLUP值均被检测到(表3和图5A),snp1691332解释9.69%的PVE。局部曼哈顿图表明A12号染色体上106.57~106.82 Mb区域有明显峰值,结合LD热图(图5B和5D)对该区间内的LD程度进行分析,将候选基因区域缩小至50 kb,在该候选区域内共注释了6个候选基因(表4)。snp1691332在GH_A12G2883内,其拟南芥的同源基因编码四肽重复样超家族蛋白;GH_A12G2884编码ARM重复超家族蛋白;GH_A12G2885编码645个氨基酸的甲基胞嘧啶结合蛋白,包含1个PHD结构域、2个环指结构域和1个SRA结构域;GH_A12G2886编码R2R3转录因子基因家族成员;GH_A12G2887编码五肽重复蛋白;GH_A12G2888编码1个与MAP激酶MAPK9相似的蛋白。单倍型分析发现携带AA单倍型的棉花品种的LP极显著高于携带GG的单倍型品种(图5C)。
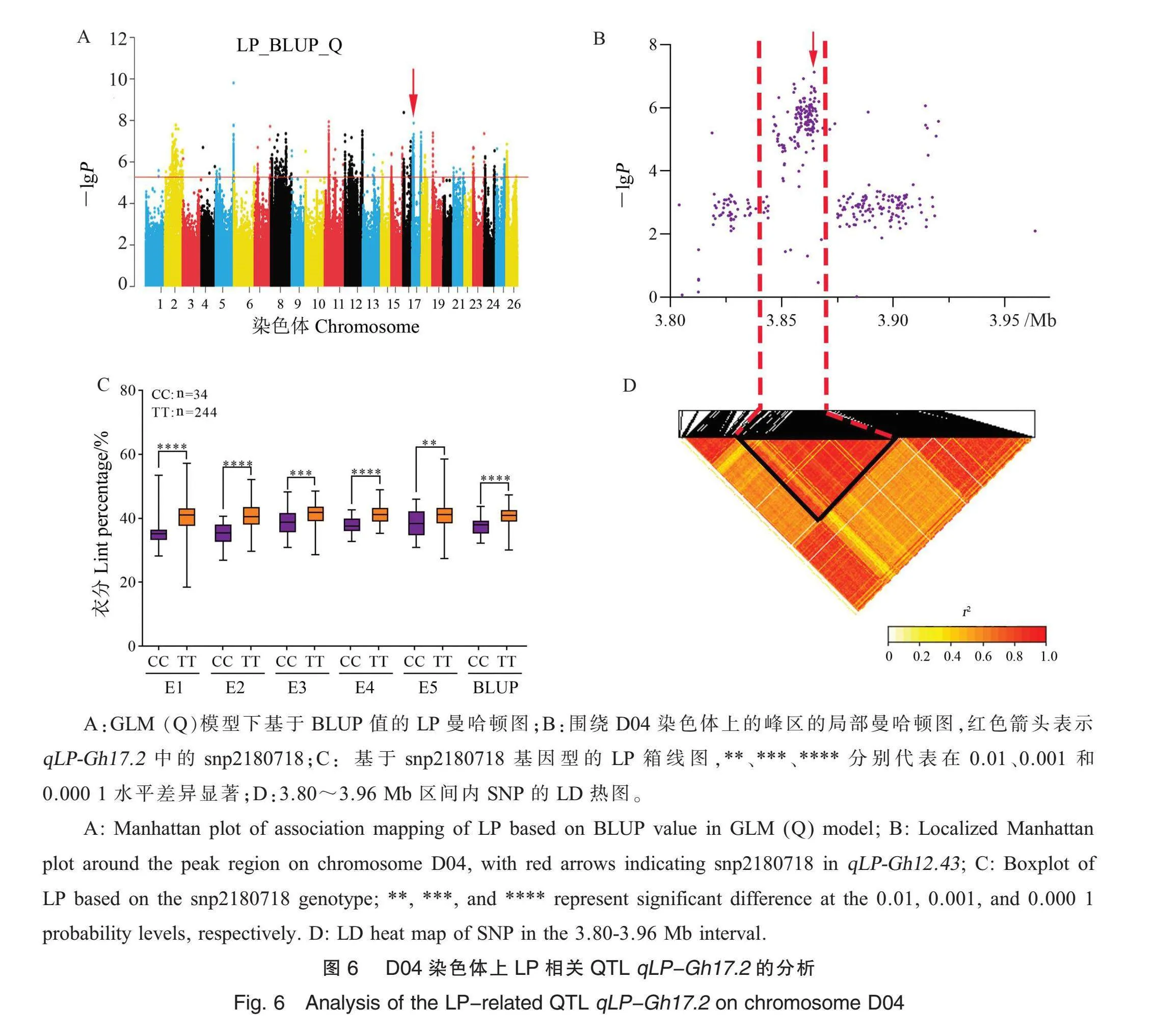
对qLP-Gh17.2进行分析,该QTL位于D04染色体上,在4种模型下均被检测到(表3和图6A),snp2180718解释10.69%的PVE。局部曼哈顿图(图6B)表明在D04号染色体上3.80~3.96 Mb区域出现明显峰值,结合热图(图6D)进一步分析该区间内的LD,将候选区域缩小至40 kb。在该候选区域内共包含4个候选基因(表4),GH_D04G0279的拟南芥同源基因编码1种IAA-氨基合成酶,在体外将天冬氨酸和其他氨基酸结合到生长素上;GH_D04G0280编码五肽重复超家族蛋白;GH_D04G0281编码肌动蛋白基因家族成员;GH_D04G0282编码GRIP卷曲蛋白(DUF1664)。单倍型分析发现携带TT单倍型的棉花品种的LP极显著高于携带CC单倍型的品种(图6C)。
对与BW相关的qBW_Gh7.5和qBW_19.5进行后续分析。qBW_Gh7.5位于A07号染色体上(图7和表3),与Sun等[20]和Ma等[16]发现的相关位点共定位(表5)。该QTL在GLM (P)和GLM (Q)模型中被检测到(表3),区间内的snp852852解释8.89%的PVE,是A07号染色体中检测到的QTL中PVE最大的位点。局部曼哈顿图(图7B)表明在A07号染色体上90. 83~91.18 Mb区域出现明显峰值,结合热图(图7D)对该区间内的LD程度进行分析,将候选区域缩小至170 kb,在该候选区域内共注释了5个候选基因(表4)。其中,GH_A07G2234的拟南芥同源基因编码RIN4-like/NOI家族成员;GH_A07G2235编码1种富含半胱氨酸的多肽,该多肽是1种在叶肉细胞中产生的分泌因子,作用于表皮以促进气孔的形成;GH_A07G2236编码天冬酰-tRNA合成酶;GH_A07G2237编码受体样蛋白激酶相关家族蛋白;GH_A07G2243编码AGC(cAMP依赖性、cGMP依赖性蛋白激酶C)激酶家族蛋白。单倍型分析发现携带GG单倍型的棉花品种的BW极显著高于携带AA单倍型的品种(图7C)。
对D06染色体上qBW-Gh19.5进行分析,该QTL在4个模型下被检测到(图8A和表3),snp2428027解释7.89%的PVE。局部曼哈顿图(图8B)表明在D06号染色体上64.22~64.56 Mb区域出现明显峰值,结合LD热图(图8D)将候选区域缩小至14 kb。该候选区域内包含3个候选基因(表4),GH_D06G2300的拟南芥同源基因编码GDSL酯酶/酰基转移酶/脂肪酶;GH_D06G2301编码花粉中参与脂滴生物形成的膜蛋白;GH_D06G2302编码1个水通道蛋白同源物。单倍型分析发现携带GG单倍型的棉花品种的BW显著高于携带AA单倍型的品种(图8C)。
3 讨论
3.1 影响GWAS结果的因素
GWAS是分析复杂性状遗传基础的有效手段之一[27],表型数据准确性、SNP标记密度、群体结构和GWAS分析方法等都会对关联分析最终结果产生很大影响。棉花的产量性状是复杂的数量性状,受多基因控制,还容易受栽培地气候、土壤肥力、田间管理、病虫害等的影响。为了减少环境对关联结果的影响,本研究对2年5个地点的LP和BW进行BLUP值的计算,以BLUP值进行关联分析,极大地减少了环境因素对遗传评估的影响,提高了结果的准确性和可靠性[28]。LD分析是GWAS的基础,受多种因素的影响,如遗传衰减、自然选择和种群结构,而群体结构被认为是影响GWAS结果的重要因素[29]。本研究中将300份陆地棉分为6个亚群,但可以看到亚群2与其他5个亚群明显分开(图2D),这可能是育种工作造成的种内渐渗[28]。研究表明所使用的群体存在较多亚群时,等位基因在基因组上的分布往往不平衡,可能造成标记与数量性状相关位点的假阳性关联,从而掩盖了真正的信号,使关联分析更加复杂[31]。PCA作协变量可以更有效地控制群体结构,同时降低过度校正的风险。所以本研究不仅使用GLM (Q)和MLM (Q+K),还使用GLM (P)和MLM (Q+ K),增加了GWAS结果的可靠性。
3.2 棉花产量性状候选基因的预测
挖掘更多的棉花产量相关基因对加速棉花高产育种具有极其重要的作用。前人已定位到许多与棉花产量性状相关的基因,如:Gh_D05G1960、Gh_D05G1965、Gh_D03G1064、Gh_D12G2354、Gh_D06G2161、Gh_A08G0716、Gh_A08G0783、Gh_A07G1389、Gh_A02G1473、Gh_A10G1765、Gh_A02G1442、Gh_D05G0313和Gh_D05G1124等都在纤维发育中起关键作用[19, 21-22, 28, 32],遗憾的是这些候选基因都未经过基因功能验证,还不能直接用于棉花产量性状的改良。
本研究在5个环境及BLUP下检测到223个QTL与LP相关,89个QTL与BW相关。91个QTL被重复检测到,88个与LP关联的QTL中9个与前人研究中共定位(表5),79个为新定位到的QTL;3个与BW关联的QTL中qBW_Gh7.5与Sun等[20]和Ma等[16]的相关研究结果共定位(表5),其余2个为新定位的QTL。
对在5个环境及BLUP下稳定检测(3次及以上)到的3个稳定的LP相关QTL进行分析,共鉴定到17个候选基因。在qLP_Gh5.18分析中,共鉴定了7个候选基因。其中,GH_A05G4228编码LUNG七跨膜受体家族蛋白,Sun等[20]在对LP的研究中定位了1个与GH_A05G4228功能相同的基因,该基因在纤维发育过程中高表达,推测GH_A05G4228可能与纤维发育有关。GH_A05G4223编码蛋白的序列与参与木质素生物合成的过氧化物酶相似,功能突变缺失表现为木质部纤维发育异常和木质素生物合成酶活性降低,推测该基因可能与纤维发育相关。在qLP_Gh12.43分析中,共发现6个候选基因,GH_A12G2887编码五肽重复超家族蛋白。TPR在棉纤维发育中的作用机制之一是与肌动蛋白形成复合物控制纤维生长[27],Zhu等[19]报道四肽重复样超家族蛋白与短纤维突变体(Ligon-lintless)表型相关,并且在开花前3 d至开花后1 d的纤维中高表达,该基因可能与纤维细胞突起有关,进而影响LP性状。GH_A12G2886编码R2R3转录因子基因家族成员,R2R3转录因子家族包括AP2/EREBP家族、MYB家族等,这些转录因子在植物的生长发育、胁迫应答等过程中发挥着关键作用[34-36]。棉花R2R3-MYB转录因子GhMYB25-like和GhMYB3均已被证明调控纤维的发育[36-37]。在qLP_Gh17.2分析中,共定位到4个候选基因,GH_D04G0279编码1种IAA-氨基合成酶,在体外将天冬氨酸和其他氨基酸结合到生长素上,目前还没有关于该基因的任何报道。但众所周知,生长素在植物生长发育过程中起着至关重要的作用,是植物生长的重要调节因子,生长素能促进植物生长,增强植物的抗逆性,提高产量和品质,故推测该基因可能通过影响棉花生长发育,从而影响LP性状;GH_D04G0280编码五肽重复超家族蛋白,棉花Gh_A03G0489编码五肽重复超家族蛋白,该基因中22 bp的缺失突变会降低纤维细胞壁厚度[38],推测GH_D04G0280可能通过调控纤维发育影响LP。
在qBW-Gh7.5区间内定位到5个候选基因,其中GH_A07G2243被重点关注,其编码AGC激酶家族蛋白,据报道其同源基因在植物中发挥多种功能,包括生长、免疫、细胞凋亡和应激反应。在水稻中AGC基因家族主要与光合作用相关,通过调控光合作用进而影响水稻产量[41]。拟南芥AGC蛋白激酶AGC1-4通过介导细胞增殖和胚胎发育进而影响种子的大小,AGC1-4过表达导致拟南芥种子变小,agc1-4突变体的种子明显大于野生型[42],因此推测GH_A07G2243可能影响棉花种子发育进而影响铃重。在qBW_
Gh19.5分析中发现3个候选基因,目前在棉花或其他作物中尚未发现关于这些基因或同源基因的报道。但我们发现GH_D06G2301编码主要在花粉中参与脂滴生物形成的膜蛋白,因此推测该基因可能通过参与花粉的形成,影响棉花的授粉,进而对棉花的产量具有一定的影响。本研究鉴定的候选基因对产量性状是否具有调控作用,还需进一步进行功能验证,才能更有效地指导棉花的育种。
4 结论
利用5个环境下对300份陆地棉的衣分和铃重及其BLUP值进行全基因组关联分析,鉴定了3个稳定的衣分相关QTL,在对应区间内挖掘到17个候选基因;鉴定了2个铃重相关QTL,挖掘到8个候选基因。
参考文献:
[1] 中国农业科学院棉花研究所. 中国棉花遗传育种学[M]. 济南:山东科学技术出版社, 2003.
Institute of Cotton Research, Chinese Academy of Agricultural Sciences. Cotton genetics and breeding in China[M]. Jinan: Shandong Science and Technology Press, 2003.
[2] Zhang Hongbin, Li Yaning, Wang Baohua, et al. Recent advances in cotton genomics[J/OL]. International Journal of Plant Genomics, 2008, 2008: 1-20[2024-03-08]. https://doi.org/
10.1155/2008/742304.
[3] 喻树迅, 范术丽, 王寒涛, 等. 中国棉花高产育种研究进展[J/OL]. 中国农业科学, 2016, 49(18): 3465-3476[2024-03-08]. https://doi.org/10.3864/j.issn.0578-1752.2016.18.001.
Yu Shuxun, Fan Shuli, Wang Hantao, et al. Progress of high-
yield cotton breeding research in China[J/OL]. Scientia Agricultura Sinica, 2016, 49(18): 3465-3476[2024-03-08]. https://doi.
org/10.3864/j.issn.0578-1752.2016.18.001.
[4] Guo Xian, Guo Yuping, Ma Jun, et al. Mapping heterotic loci for yield and agronomic traits using chromosome segment introgression lines in cotton[J/OL]. Journal of Integrative Plant Biology, 2013, 55(8): 759-774[2024-03-08]. https://doi.org/10.1111/
jipb.12054.
[5] Liu Rezhong, Ai Nijiang, Zhu Xinxia, et al. Genetic analysis of plant height using two immortalized populations of “CRI12×J8891” in Gossypium hirsutum L.[J/OL]. Euphytica, 2014, 196(1): 51-61[2024-03-08]. https://doi.org/10.1007/s10681-013-
1013-0.
[6] Shao Qianshun, Zhang Fengjiao, Tang Shiyi, et al. Identifying QTL for fiber quality traits with three upland cotton (Gossypium hirsutum L.) populations[J/OL]. Euphytica, 2014, 198(1): 43-58[2024-03-08]. https://doi.org/10.1007/s10681-014-1082-8.
[7] Yu Jiwen, Zhang Ke, Li Shuaiyan, et al. Mapping quantitative trait loci for lint yield and fiber quality across environments in a Gossypium hirsutum × Gossypium barbadense backcross inbred line population[J/OL]. Theoretical and Applied Genetics, 2013, 126: 275-287[2024-03-08]. https://doi.org/10.1007/s00122-012-
1980-x.
[8] Ning Zhiyuan, Zhao Rui, Chen Hong, et al. Molecular tagging of a major quantitative trait locus for broad-spectrum resistance to Verticillium wilt in upland cotton Cultivar prema[J/OL]. Crop Science, 2013, 53(6): 2304-2312[2024-03-08]. https://doi.org/
10.2135/cropsci2012.12.0694.
[9] Islam M S, Zeng LH, Thyssen G N, et al. Mapping by sequencing in cotton (Gossypium hirsutum) line MD52ne identified candidate genes for fiber strength and its related quality attributes[J/OL]. Theoretical and Applied Genetics, 2016, 129: 1071-1086[2024-
03-08]. https://doi.org/10.1007/s00122-016-2684-4.
[10] 张雁明, 邢国芳, 刘美桃, 等. 全基因组关联分析: 基因组学研究的机遇与挑战[J]. 生物技术通报, 2013(6): 1-6.
Zhang Yanming, Xing Guofang, Liu Meitao, et al. Genome wide association study: opportunities and challenges in genomic research[J]. Biotechnology Bulletin, 2013(6): 1-6.
[11] Shao Panxia, Peng Yabin, Wu Yuanlong, et al. Genome-wide association study and transcriptome analysis reveal key genes controlling fruit branch angle in cotton[J/OL]. Frontiers in Plant Science, 2022, 13: 9988647[2024-03-08]. https://doi.org/10.3389/fpls.2022.988647.
[12] Ye Yulu, Wang Peilin, Zhang Man, et al. UAV-based time series phenotyping reveals the genetic basis of plant height in upland cotton[J/OL]. The Plant Journal, 2023, 115(4): 937-951[2024-03-08]. https://doi.org/10.1111/TPJ.16272.
[13] Fang Lei, Wang Qiong, Hu Yan, et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits[J/OL]. Nature Genetics, 2017, 49(7): 1089-1098[2024-03-08]. https://doi.org/10.1038/ng.3887.
[14] Sarfraz Zareen, Iqbal Muhammad Shahid, Geng Xiaoli, et al. GWAS mediated elucidation of heterosis for metric traits in cotton (Gossypium hirsutum L.) across multiple environments[J/OL]. Frontiers in Plant Science, 2021, 12: 565552[2024-03-
08]. https://doi.org/10.3389/fpls.2021.565552.
[15] Sun Zhengwen, Wang Xingfen, Liu Zhengwen, et al. Genome-
wide association study discovered genetic variation and candidate genes of fibre quality traits in Gossypium hirsutum L.[J/OL]. Plant Biotechnology Journal, 2017, 15(8): 982-996[2024-03-08]. https://doi.org/10.1111/pbi.12693.
[16] Ma Zhiying, He Shoupu, Wang Xingfen, et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield[J/OL]. Nature Genetics, 2018, 50(6): 803-813[2024-03-08]. https://doi.org/10.1038/s41588-018-0119-7.
[17] Ulloa M, De Santiago L M, Hulse-Kemp A M, et al. Enhancing upland cotton for drought resilience, productivity, and fiber quality: comparative evaluation and genetic dissection[J/OL]. Molecular Genetics and Genomics, 2020, 295: 155-176[2024-
03-08]. https://doi.org/10.1007/s00438-019-01611-6.
[18] 韩贝, 王旭文, 李保奇, 等. 陆地棉种质资源抗旱性状的关联分析[J/OL]. 作物学报, 2021, 47(3): 438-450[2024-03-08]. https://doi.org/10.3724/SP.J.1006.2021.04063.
Han Bei, Wang Xuwen, Li Baoqi, et al. Association analysis of drought tolerance traits of upland cotton accessions (Gossypium hirsutum L.)[J/OL]. Acta Agronomica Sinica, 2021, 47(3): 438-450[2024-03-08]. https://doi.org/10.3724/SP.J.1006.2021.
04063.
[19] Zhu Guozhong, Hou Sen, Song Xiaohui, et al. Genome-wide association analysis reveals quantitative trait loci and candidate genes involved in yield components under multiple field environments in cotton (Gossypium hirsutum)[J/OL]. BMC Plant Biology, 2021, 21(1): 250-250[2024-03-08]. https://doi.org/10.1186/S12870-021-03009-2.
[20] Sun Zhengwen, Wang Xingfen, Liu Zhengwen, et al. A genome-
wide association study uncovers novel genomic regions and candidate genes of yield-related traits in upland cotton[J/OL]. Theoretical and Applied Genetics, 2018, 131: 2413-2425[2024-
03-08]. https://doi.org/10.1007/s00122-018-3162-y.
[21] Song Chengxiang, Li Wei, Pei Xiaoyu, et al. Dissection of the genetic variation and candidate genes of lint percentage by a genome-wide association study in upland cotton[J/OL]. Theoretical and Applied Genetics, 2019, 132: 1991-2002[2024-
03-08]. https://doi.org/10.1007/s00122-019-03333-0.
[22] Wang Yuanyuan, Guo Xinlei, Xu Yanchao, et al. Genome-wide association study for boll weight in Gossypium hirsutum races[J/OL]. Functional amp; Integrative Genomics, 2023, 23(4): 331[2024-03-08]. https://doi.org/10.1007/S10142-023-01261-3.
[23] Huang Cong, Nie Xinhui, Shen Chao, et al. Population structure and genetic basis of the agronomic traits of upland cotton in China revealed by a genome-wide association study using high-density SNPs[J/OL]. Plant Biotechnology Journal, 2017, 15(11): 1374-1386[2024-03-08]. https://doi.org/10.1111/pbi.
12722.
[24] 杜雄明, 周忠丽. 棉花种质资源描述规范和数据标准[M]. 北京: 中国农业出版社, 2005.
Du Xiongming, Zhou Zhongli. Cotton germplasm resources description specification and data standard[M]. Beijing: China Agricultural Press, 2005.
[25] Zhou Xiang, Stephens M. Genome-wide efficient mixed-model analysis for association studies[J/OL]. Nature Genetics, 2012, 44(7): 821-824[2024-03-08]. https://doi.org/10.1038/ng.2310.
[26] Li MX, Yeung J M Y, Cherny S S, et al. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets[J/OL]. Human Genetics, 2012, 131(5): 747-756[2024-03-08]. https://doi.org/10.1007/s00439-011-
1118-2.
[27] Yasir M, Kanwal H H, Hussain Q, et al. Status and prospects of genome-wide association studies in cotton[J/OL]. Frontiers in Plant Science, 2022, 13: 1019347[2024-03-08]. https://doi.org/
10.3389/FPLS.2022.1019347.
[28] 王娟, 马晓梅, 周小凤, 等. 棉花产量构成因素性状的全基因组关联分析[J/OL]. 中国农业科学, 2022, 55(12): 2265-2277[2024-03-08]. https://doi.org/10.3864/j.issn.0578-1752.2022.
12.001.
Wang Juan, Ma Xiaomei, Zhou Xiaofeng, et al. Genome-wide association analysis of yield component traits in cotton[J/OL]. Scientia Agricultura Sinica, 2022, 55(12): 2265-2277[2024-
03-08]. https://doi.org/10.3864/j.issn.0578-1752.2022.12.001.
[29] Lon R C, Lyle J P. Population stratification and spurious allelic association[J/OL]. The Lancet, 2003, 361(9357): 598-604[2024-
03-08]. https://doi.org/10.1016/S0140-6736(03)12520-2.
[30] Zhao N, Wang W R, Grover C E, et al. Genomic and GWAS analyses demonstrate phylogenomic relationships of Gossypium barbadense in China and selection for fibre length, lint percentage and Fusarium wilt resistance[J/OL]. Plant Biotechnology Journal, 2021, 20(4): 691-710[2024-03-08]. https://doi.org/
10.1111/pbi.13747.
[31] 马艳明, 娄鸿耀, 张胜军, 等. 新疆冬小麦品种资源主要产量性状全基因组关联分析[J/OL]. 中国农业科学, 2023, 56(18): 3487-3499[2024-03-08]. https://doi.org/10.3864/j.issn.0578-
1752.2023.18.001.
Ma Yanming, Lou Hongyao, Zhang Shengjun, et al. Genome-
wide association analysis of major yield traits of winter wheat varietal resources in Xinjiang[J/OL]. Scientia Agricultura Sinica, 2023, 56(18): 3487-3499[2024-03-08]. https://doi.org/10.3864/
j.issn.0578-1752.2023.18.001.
[32] Su Junji, Wang Caixiang, Ma Qi, et al. An RTM-GWAS procedure reveals the QTL alleles and candidate genes for three yield-related traits in upland cotton[J/OL]. BMC Plant Biology, 2020, 20(1): 416-416[2024-03-08]. https://doi.org/10.1186/
s12870-020-02613-y.
[33] Zubair I, Muhammad S I, Salman A, et al. Genome-wide association study reveals novel genes on different chromosomal positions regulating boll weight in upland cotton (Gossypium hirsutum L.)[J/OL]. Genetic Resources and Crop Evolution, 2023, 71(2): 785-799[2024-03-08]. https://doi.org/10.1007/
s10722-023-01657-x.
[34] Liu Chunxiao, Zhang Tianzhen. Expansion and stress responses of the AP2/EREBP superfamily in cotton[J/OL]. BMC Genomics, 2017, 18(1): 118[2024-03-08]. https://doi.org/10.1186/s12864-
017-3517-9.
[35] Gates D J, Strickler S R, Mueller L A, et al. Diversification of R2R3-MYB transcription factors in the tomato family solanaceae[J/OL]. Journal of Molecular Evolution, 2016, 83(1/2): 26-37[2024-03-08]. https://doi.org/10.1007/s00239-016-9750-z.
[36] Shangguan X, Yang Q, Wu X, et al. Function analysis of a cotton R2R3 MYB transcription factor GhMYB3 in regulating plant trichome development[J/OL]. Plant Biology, 2021, 23(6): 1118-1127[2024-03-08]. https://doi.org/10.1111/PLB.13299.
[37] Hu Haiyan, He Xin, Tu Lili, et al. GhJAZ2 negatively regulates cotton fiber initiation by interacting with the R2R3-MYB transcription factor GhMYB25-like[J/OL]. The Plant Journal, 2016, 88(6): 921-935[2024-03-08]. https://doi.org/10.1111/tpj.13273.
[38] Thyssen G N, Fang D D, Zeng L H, et al. The immature fiber mutant phenotype of cotton (Gossypium hirsutum) is linked to a 22-bp frame-shift deletion in a mitochondria targeted pentatricopeptide repeat gene[J/OL]. G3: Genes, Genomes, Genetics, 2016, 6(6): 1627-1633[2024-03-08]. https://doi.org/10.1534/
g3.116.027649.
[39] Nazir M F, Jia Y H, Ahmed H, et al. Genomic insight into differentiation and selection sweeps in the improvement of upland cotton[J/OL]. Plants, 2020, 9(6): 711[2024-03-08]. https://doi.org/10.3390/plants9060711.
[40] 侯森. 陆地棉耐旱及产量相关性状的全基因组关联分析与优异等位变异挖掘[D]. 南京: 南京农业大学, 2021.
Hou Sen. Genome-wide association analysis and superior allelic variation mining for drought tolerance and yield-related traits in land cotton[D]. Nanjing: Nanjing Agricultural University, 2021.
[41] Jiang Yifei, Liu Xuhui, Zhou Mingao, et al. Genome-wide identification of the AGC protein kinase gene family related to photosynthesis in rice (Oryza sativa)[J/OL]. International Journal of Molecular Sciences, 2022, 23(20): 12557-12557[2024-03-08]. https://doi.org/10.3390/IJMS232012557.
[42] Zhang Yuying, Yao Wangjinsong, Wang Fang, et al. AGC protein kinase AGC1-4 mediates seed size in Arabidopsis[J/OL]. Plant Cell Reports, 2020, 39(6): 1-13[2024-03-08]. https://doi.org/10.1007/s00299-020-02533-z.
(责任编辑:王国鑫 责任校对:王小璐)
猜你喜欢
中国棉花加工(2023年4期)2023-09-27 11:28:58
中国棉花(2023年1期)2023-05-30 10:48:04
江西农业学报(2020年8期)2020-09-02 06:54:30
天津农业科学(2020年4期)2020-07-07 09:31:48
棉花科学(2020年2期)2020-06-01 18:40:32
中国纤检(2019年5期)2019-12-09 16:06:20
中国棉花(2018年7期)2018-08-29 00:54:08
中国棉花加工(2018年1期)2018-04-24 07:25:48
中国种业(2016年6期)2016-07-27 11:58:34
安徽农学通报(2016年7期)2016-05-03 02:11:14
