
大肠杆菌合成烟酰胺单核苷酸途径构建及发酵工艺研究
2023-12-29 04:56程琳龚劲松MichaelHALL苏畅徐国强许正宏1史劲松1
食品与发酵工业 2023年24期
程琳,龚劲松,Michael HALL,苏畅,徐国强,许正宏1,,史劲松1,*
1(江南大学 生物工程学院,糖化学与生物技术教育部重点实验室,江苏 无锡,214122) 2(江南大学 生命科学与健康工程学院,江苏 无锡,214122) 3(江南大学 粮食发酵与食品生物制造国家工程研究中心,江苏 无锡,214122) 4(Seragon Biosciences, Inc., California Irvine, 92618)
烟酰胺单核苷酸(nicotinamide mononucleotide,NMN)是烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,NAD+)的前体[1],NAD+是生物体内重要的调节辅酶,参与调节能量代谢、细胞凋亡和增殖[2-4]等诸多通路。补充NAD+可预防心血管疾病、修复肾脏损伤和保护视神经等,但NAD+因分子质量大难以直接进入细胞,而补充其前体NMN更能有效提高人体NAD+水平[5]。已有研究表明,补充NMN对机体的心脑组织损伤、代谢性疾病、衰老及其相关老化退行性疾病等均有较好的预防、治疗和修复作用[6-8],并有望应用于治疗肥胖相关的肝病、帕金森和阿尔茨海默症,改善组织功能[3,9-10]。
目前合成NMN的方法主要有化学合成法、化学-生物酶法和微生物发酵法,化学法合成路线复杂、试剂昂贵、总收率相对较低[11];化学-生物酶法一方面需要高活性生物酶,同时采用的底物仍需要复杂的化学工艺支持,产品的绿色性、安全性有待提升,因此,近年来微生物发酵法生产NMN逐步受到重视[12]。NMN代谢合成途径主要有3条,a)NAD+补救途径,以5-磷酸核糖-1-焦磷酸(5-phosphoribose 1-pyrophosphate,PRPP)和烟酰胺(nicotinamide,NAM)为底物通过烟酰胺磷酸核糖转移酶(Nampt,EC 2.4.2.12)催化生成NMN[13-14];b)NAD+替代补救途径,以烟酸核糖激酶(NPK EC 2.7.1.173)催化烟酰胺核糖(nicotinamide riboside,NR)和ATP生成NMN[15]。上述两个途径主要存在于哺乳动物生物体中;c)通过Preiss-Handler途径,以磷酸核糖基转移酶(NaPRT,EC 2.4.2.11)催化PRPP合成烟酸单核苷酸(niacin mononucleotide,NaMN),而来源于土拉弗朗西斯菌中的NMN合成酶则可以将NaMN转化为NMN[16]。
由于替代补救途径中的NR中间代谢物价格较为昂贵[5],是规模化制备NMN的一大障碍,因此目前微生物发酵法生产NMN大多采用NAD+补救途径,通过异源筛选高效的NAMPT在细菌体内构建NAD+补救途径[17]。2018年首次报道了以大肠杆菌为宿主表达来源于杜克雷嗜血杆菌的NAMPT用于合成NMN,胞外质量浓度为15.4 mg/L[18],此后的研究者通过对PRPP生物合成途径酶进行过表达来增加PRPP的供应,或者通过增加底物NAM进胞、产物出胞的能力以提升NMN的积累[19],并通过染色体整合表达技术,显著提高NAD+的产量和合成稳定性[20]。
本研究以大肠杆菌BL21(DE3)为出发宿主,通过异源表达Nampt构建NMN的合成途径,在增强表达合成前体PRPP的途径酶的同时,敲除NMN代谢降解相关基因pncC、ushA以弱化NMN的降解,并敲除NAD+衍生物调控基因nadR,探讨采用多种代谢策略提高NMN的发酵产量。
1 材料与方法
1.1 材料
1.1.1 菌株、质粒和引物
用于质粒构建的克隆宿主为大肠杆菌(Escherichiacoli)JM109,表达宿主为E.coliBL21(DE3),用于大肠杆菌体系的表达质粒为pCDFDuet。实验所用及构建的菌株、质粒如表1所示,引物序列参见表2,引物合成由天霖生物有限公司完成。

表1 本研究所用菌株与质粒Table 1 Strains and plasmids used in the study
1.1.2 实验试剂
DNA聚合酶及Marker,TaKaRa公司;同源重组酶、DNA 纯化及质粒提取试剂盒,诺维赞公司。
1.1.3 培养基
LB培养基(g/L):蛋白胨10,酵母粉5,氯化钠10,自然pH。
种子培养基(g/L):胰蛋白胨20,酵母粉5,NaCl 0.50,KCl 0.19,MgCl20.95,葡萄糖3.60,pH 7.0;微量元素2 mL/L。
发酵培养基(g/L):胰蛋白胨20,酵母粉5,NaCl 0.50,KCl 0.19,MgCl20.95,葡萄糖3.60,pH 7.0;微量元素2 mL/L。

表2 本研究所用引物Table 2 Primers used in the study
补料培养基(g/L):葡萄糖400,胰蛋白胨100,酵母粉80,MgSO414;微量元素14 mL/L。
微量元素溶液(g/L):FeCl3·6H2O 6,ZnSO4·7H2O 0.58,CaCl20.20,CuCl2·2H2O 0.20,MnSO4·H2O 0.30,EDTA 0.50。
1.2 重组质粒的构建
以pCDFDuet-Nampt质粒构建为例,以大肠杆菌基因组为模板,扩增获得基因片段(引物如表1所示),选用诱导型质粒pCDFDuet,用限制性核酸内切酶ndeI、kpnI双酶切后,将其与目的基因进行同源重组连接,然后将重组产物转入JM109感受态细胞,在含有链霉素的LB平板上筛选阳性克隆,得重组质粒。
1.3 CRISPR/Cas9系统敲除大肠杆菌基因
使用CRISPR/Cas9双质粒系统敲除pncC、nadR、ushA基因,以敲除pncC为例,将含有Cas9编码基因的pCas9质粒化转入表达宿主中,然后制备成电转感受态。
通过网站CHOPCHOP设计得到sgRNA序列,采用引物pncC-sgRNA-F和pncC-sgRNA-R将打靶质粒pTarget进行PCR反向扩增,引入20 bp的sgRNA序列,随后进行转化提取质粒,获得基因敲除打靶质粒pTarget-pncC。
选取目标基因上下游各500 bp,采用引物分别扩增基因上游同源臂、基因下游同源臂,然后利用重叠延伸PCR技术获得1 000 bp的上下游同源臂,构建获得修复模板。
将敲除质粒和修复模板电转入感受态中,验证敲除正确后,诱导pCas9质粒定位pTarget质粒上的sgRNA,从而消除pTarget质粒;借助pCas9质粒是温敏型质粒的特性,在42 ℃培养12~15 h,消除pCas9质粒,得到BL21(DE3)ΔpncC。
1.4 基因表达的鉴定
挑取单克隆于10 mL的LB培养基中,37 ℃培养12 h。再将上述菌液按1%接种量转接30 mL发酵培养基中培养,37 ℃培养至OD600值为0.6~0.8时,添加终浓度为0.5 mmol/L的IPTG,于25 ℃下诱导8~12 h,取样,离心,菌体经超声破碎,取上清液,采用SDS-PAGE检测目标蛋白。
1.5 重组菌株的发酵研究
种子培养:从活化平板上挑取单菌落接种至10 mL LB液体培养基中,添加终质量浓度100 μg/mL链霉素,37 ℃、220 r/min 振荡培养10~12 h。
摇瓶发酵:种子液按1%接种量转接30 mL发酵培养基中,37 ℃培养至OD600值为0.6~0.8时,添加终浓度为0.5 mmol/L的IPTG,于25 ℃下诱导8~12 h,测定NMN积累量。
5 L罐发酵:将种子培养液按照10%(体积分数)接种量转接于5 L发酵罐,初始培养温度37 ℃,pH值控制在7.0±0.2,通气量设置为0.18 m3/h,转速关联溶氧量自动调节,溶氧量维持在10%~30%,装液量3 L,菌体培养至OD600值为8~10时,加入终浓度为0.5 mmol/L的IPTG进行诱导,表达培养温度为25 ℃,定期取样测定生物量和NMN产量。
1.6 检测方法
NMN检测方法:取发酵液1 mL,离心取上清液即为发酵上清液;菌体经磷酸钠缓冲溶液(phosphate buffer solution,PBS)洗涤后超声破碎,离心后取上清液即为胞内上清液。样品测定前通过孔径0.22 μm的滤膜过滤,然后用高效液相色谱进行检测。液相色谱柱为Diamonsil 5 μL C18 250 mm×4.6 mm;流动相A:100%甲醇,流动相B:5 mmol/L四丁基氢氧化的5%甲醇水溶液。流速为0.7 mL/min;检测波长为254 nm,进样量为10 μL,柱温30 ℃。洗脱方案:0~20 min,100% A;20~25 min,40% A、60% B;25~35 min,20% A、80% B;35~40 min,100% A。
发酵液中葡萄糖含量测定:利用西尔曼生物生物传感器进行分析。
2 结果与分析
2.1 构建NMN合成途径
大多数细菌通过NAD+替代补救途径即烟酸核苷激酶从NR和ATP生成,而在哺乳动物生物体中,可以通过NAD+补救途径即通过Nampt以PRPP 和NAM为底物生成NMN,因NR较为昂贵,目前微生物发酵法生产NMN大多采用NAD+补救途径,通过筛选异源Nampt生产NMN[21]。本研究以大肠杆菌为表达体系,以松噬几丁质菌来源的Nampt基因片段构建NMN合成途径(图1-a),首先,通过PCR扩增获得Nampt基因片段,将目的片段和双酶切载体进行同源重组、连接转化获得转化子。Nampt基因片段长度约为1 400 bp,菌落PCR验证,其目的条带大小与预期一致,提取质粒送样测序,确认重组质粒构建成功,转入表达宿主,获得了重组菌B1。将B1菌株发酵10 h后,收集菌体进行超声破碎,取上清液进行SDS-PAGE分析(图1-b)。
Nampt基因表达的目的蛋白理论分子质量为52.7 kDa,其SDS-PAGE目的条带与理论值基本一致。将重组菌株进行摇瓶发酵,生成的NMN产量为12.1 mg/L,而对照组未测出NMN。

a-pcDFDuet-Nampt重组质粒图谱;b-重组蛋白Nampt的 SDS-PAGE电泳分析图( M-蛋白Marker;1-空白对照, 含空载质粒的BL21(DE3);2-发酵上清液;3-破壁上清液)图1 重组蛋白Nampt的构建和验证Fig.1 Construction and validation of the recombinant protein Nampt
2.2 增强PRPP途径
合成NMN所需的前体之一是PRPP,PRPP的生物合成途径与细胞生长竞争碳通量[22],因此提高NMN产量的重要策略之一就是增强PRPP的供应。根据代谢网络,PRPP是细胞重要代谢中间体,涉及嘌呤核苷酸、嘧啶核苷酸、色氨酸、组氨酸等多种代谢产物合成。PRPP主要通过磷酸戊糖途径生成,因此可将相关基因进行过表达来增强PRPP的供应,本研究构建了质粒pcDFDuet-zwf-pgl-gnd-rpiA-rpiB-prs-Nampt,表示为pcDFDuet-zwf→Nampt,如图2-a所示,将该质粒导入获得重组菌B2。通过SDS-PAGE对6-磷酸葡萄糖脱氢酶(zwf)、6-磷酸葡萄糖内酯酶(pgl)、6-磷酸葡萄糖脱氢酶(gnd)、戊糖磷酸异构酶(rpiA、rpiB)、PRPP合酶(prs)的表达情况进行验证分析,结果如图2-b所示。zwf、pgl、gnd、rpiA、rpiB、prs基因表达的目的蛋白理论分子质量分别为55.7、36.3、51.5、22.9、16.1、33.9 kDa,这6个蛋白的SDS-PAGE条带与理论相符,大小一致,说明均已成功表达。经发酵验证,其NMN产量为31.7 mg/L,是未强化的工程菌产量2.6倍,但是摇瓶OD600最大值为2.7,低于空白对照(OD600=3.1),推测单质粒共表达多个酶对宿主的负担较大,影响了生长。
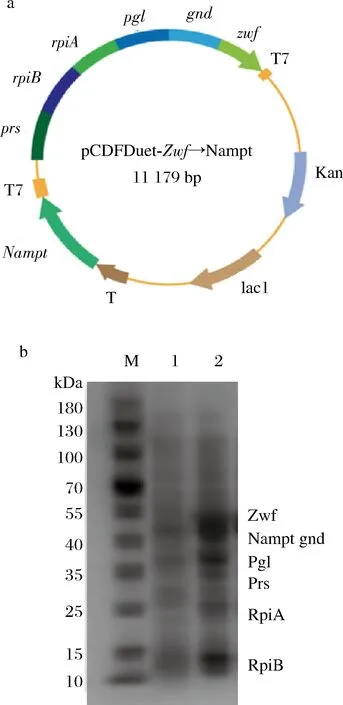
a-pcDFDuet-zwf→Nampt重组质粒示意图谱; b-重组蛋白SDS-PAGE电泳图 (M-蛋白质Marker;1-空白对照;2-破壁上清液)图2 重组质粒pcDFDuet-zwf→Nampt的构建和验证Fig.2 Construction and validation of the recombinant plasmid pcDFDuet-zwf→Nampt
2.3 NMN下游代谢途径的基因敲除
弱化目的产物的降解是一种有效提高产量的代谢改造方式。大肠杆菌中NMN的下游代谢主要是进行NR、NAD的合成(图3),如需在大肠杆菌有效积累NMN,必须对其代谢途径进行阻断。为此,本研究利用CRISPR/Cas9系统精准敲除了NMN代谢过程中关键基因pncC、ushA和NAD衍生物调控基因nadR,获得有利于积累NMN的基因工程菌。

图3 大肠杆菌利用葡萄糖和烟酰胺合成NMN的代谢途径Fig.3 Synthesis of NMN by E.coli using glucose and niacinamide注:红色箭头表示利用质粒进行过表达,pncC、ushA、nadR为 本研究敲除基因。
2.3.1 NMN氨基水解酶的失活
大肠杆菌中的NMN氨基水解酶(PncC)可将烟酰胺单核苷酸脱氨为烟酸单核苷酸,本文在加强途径酶表达的基础上敲除NMN氨基水解酶,按照1.3节中的方法对该基因进行敲除。构建修复模板(图4-a)和打靶质粒(图4-b),验证结果如图4-c所示。NMN氨基水解酶基因大小为498 bp,若敲除成功,则条带大小应约为1 200 bp,将条带大小正确的扩增片段进一步测序验证。测序结果显示与同源臂序列一致,表明pncC基因敲除成功。将该工程菌B3进行摇瓶发酵,以HPLC检测产量如图5所示,发酵结果表明该基因敲除后未对菌株的生长造成影响,而产量提升至44.3 mg/L。
2.3.2 5-核苷酸酶的失活
如图3大肠杆菌中代谢网络所示,5-核苷酸酶(UshA)可催化NMN变成烟酰胺核糖苷NR。为进一步提高NMN的产量,在敲除NMN氨基水解酶的基础上继续敲除5-核苷酸酶。5-核苷酸酶基因大小为1 653 bp,敲除后约为1 200 bp,验证结果如图4-d所示,将该改造菌B4进行摇瓶发酵,结果如图6所示,表明敲除5-核苷酸酶未对菌株的生长造成影响,NMN产量则提升至51.6 mg/L。

a-修复模板扩增产物(泳道1、2为扩增条带);b-pTargetF质粒 扩增产物(泳道1、2为扩增条带);c-pncC基因敲除验证(泳道1为 对照菌株,泳道2为pncC基因敲除的菌株);d-pncC、ushA基因敲除 验证(泳道1、3为对照菌株,泳道2、4分别为pncC、ushA基因敲除的 菌株);e-pncC、ushA、nadR基因敲除验证(泳道1、3、5为对照菌株, 泳道2、4、6分别为pncC、ushA、nadR基因敲除的菌株)图4 基因敲除菌株的电泳验证图Fig.4 Verification electropherogram of gene knockout strains注:M-DNA marker。
2.3.3 NAD衍生物合成调控酶的失活
大肠杆菌中存在一种调控酶(NadR),具有调控NAD衍生物合成的能力,这是一种负反馈调控,当NAD衍生物累积一定量时会抑制NMN合成关键酶Nampt的表达,不利于NMN的积累。NadR调控酶基因大小为1 233 bp,未敲除成功的条带大小约为2 400 bp,成功敲除的约为1 200 bp(图4-e)。将该改造菌进行摇瓶发酵,结果表明,敲除未对菌株的生长造成明显影响,NMN能够得到有效积累,产量提升至60 mg/L,为初始表达产量的4.95倍。
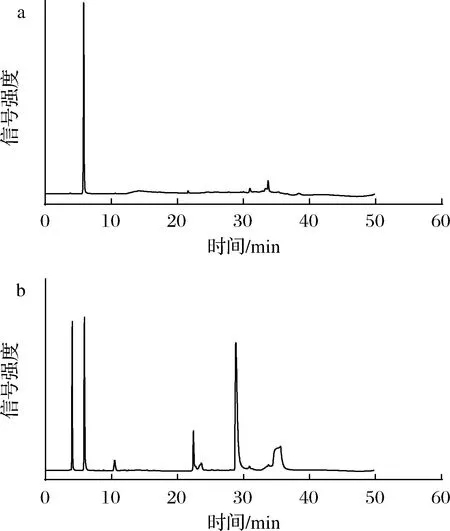
a-标准品的HPLC检测图谱;b-发酵破碎上清液的HPLC检测图谱图5 NMN的标准品及发酵液HPLC检测图谱Fig.5 HPLC detection results of NMN standards and fermentation broth

a-最大OD600测定值;b-发酵结束NMN终产量图6 重组菌株的摇瓶发酵参数Fig.6 Shaker fermentation parameters of recombinant strains注:B0-空白对照菌株;B1-BL21(DE3)pCDFDuet-Nampt;B2-BL21(DE3) pCDFDuet-zwf→Nampt;B3-B2ΔpncC;B4-B3ΔushA;B5-B4ΔnadR。
2.4 小罐发酵工艺研究
为了进一步探究工程菌的发酵合成潜力,在5 L小罐上进行发酵工艺研究。考察了两种发酵策略对菌体生长及产量的影响(图7)。第一种如图7-a所示,当葡萄糖消耗完,开始流加葡萄糖和烟酰胺,葡萄糖作为菌体生长的碳源的同时也是合成NMN的前体物质,发酵过程中控制流加速度使葡萄糖和烟酰胺维持在较低浓度,最高NMN产量为290.7 mg/L;第二种如图7-b所示,当葡萄糖消耗完后,开始补料培养基和烟酰胺,同样控制流加速度使葡萄糖和烟酰胺维持在较低浓度,避免高浓度导致的高渗透压对菌体生长带来不利影响,在36 h时产量最高达到390.1 mg/L,是摇瓶水平的6.5倍,OD600值最高为16.4。相比而言,第二种补料策略较好,可能是培养基的成分更丰富,能充分满足菌体生长的需要,进而影响NMN的合成。

a-流加葡萄糖、烟酰胺的发酵过程参数;b-补料培养基、 葡萄糖、烟酰胺的发酵过程参数图7 重组菌在5 L发酵罐中发酵生产NMN的过程曲线Fig.7 Process curve of NMN production by the recombinant strain in 5 L fermenter
3 结论
本研究以大肠杆菌BL21(DE3)作为表达宿主,通过引入外源松噬几丁质菌的Nampt基因,在大肠杆菌中成功实现了NMN的生物合成。通过强化表达大肠杆菌自身的zwf、pgl、gnd、ripA、ripB、prs基因,增强了PRPP途径的合成能力。为了使大肠杆菌有效积累产物NMN,敲除了NMN的代谢基因pncC、ushA以及调控因子nadR。进而通过重组菌株的发酵工艺的初步研究,使菌株的摇瓶最高产量为60 mg/L,5 L发酵罐产量达到390.1 mg/L。后期研究将基于该工程菌株,重点开展关键合成基因的性能提升、前体物质的进胞、产物出胞以及基于细胞能量水平的优化工作,进一步探索高产NMN的工程菌株构建策略。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
食品科学(2018年10期)2018-05-23
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年9期)2015-03-01
西华师范大学学报(自然科学版)(2015年3期)2015-02-27
西南军医(2015年6期)2015-01-23
药学与临床研究(2014年3期)2014-03-06
癌变·畸变·突变(2014年2期)2014-03-01
