
MgO活性对碱矿渣砂浆抗碳化性能的影响
2023-12-28 13:17:46梁咏宁吴明勤郑昊王耀季韬
福州大学学报(自然科学版) 2023年6期
梁咏宁, 吴明勤, 郑昊, 王耀, 季韬
(1. 福州大学先进制造学院, 福建 泉州 362251; 2. 福州大学土木工程学院, 福建 福州 350108; 3. 中建海峡建设发展有限公司, 福建 福州 350015)
0 引言
碱矿渣水泥(alkali-slag cement, ASC)是一种采用碱性激发剂和高炉矿渣混合制成的胶凝材料[1]. 与硅酸盐水泥相比, ASC具有需水量小、 水化热低、 强度高、 节能环保等优点[2-3], 但同时也存在抗碳化性能差、 易开裂等缺点[4-5]. 碳化会降低混凝土的碱性, 使钢筋更易发生锈蚀, 从而破坏钢筋混凝土结构的安全性[6-7]. Palacios等[8]认为ASC的抗碳化性能较硅酸盐水泥差的主要原因, 是ASC的水化产物中缺少可以与CO2先行反应的缓冲物质(如硅酸盐水泥中的水化产物Ca(OH)2), 导致决定强度的水化硅酸钙(C-S-H)凝胶直接与CO2接触并反应. 何娟等[9]认为孔隙溶液中存在较多的碱(Na+和K+)、 混凝土的干燥收缩较大是ASC碳化严重的重要原因.
为提高ASC的抗碳化性能, Mohammad等[10]用偏高岭土替代矿渣来制备ASC, 结果表明, 随着偏高岭土掺入量的增加, ASC的孔隙率下降, CO2向ASC内部的扩散受到抑制, 使得ASC的抗碳化性能得到提高. Busra等[11]将MgO和偏高岭土按一定比例复合, 并等质量替代ASC中的矿渣. 与偏高岭土相比, MgO能更好地提高ASC的抗碳化性能. 这主要是因为MgO可以稳定ASC中的无定形CaCO3, 从而抑制C-S-H凝胶的进一步脱钙, 提高ASC的抗碳化性能[12]. 郑昊等[13]分别用MgO和CaO替代部分矿渣, 制备碱矿渣混凝土. 研究表明, 在加速碳化条件下, MgO和CaO都可以提高碱矿渣混凝土的抗碳化性能. 但是, MgO对碱矿渣混凝土的抗压强度和抗碳化性能的提升效果明显优于CaO. 综上所述, 在ASC中掺入MgO可有效改善ASC的抗碳化性能. 但是, 目前关于MgO活性对ASC抗碳化性能影响方面的研究鲜有报道.
本研究分别用3种不同活性的MgO替代矿渣来制备碱矿渣砂浆(alkali-activated slag mortar, AASM), 研究碳化前AASM的吸水率、 抗压强度的变化规律和不同加速碳化龄期下AASM的碳化深度(d碳化)、 抗压强度(fc)和抗压强度保留率, 并进一步分析加速碳化前后MgO活性对AASM的水化和碳化产物、 孔结构的影响, 揭示MgO活性对AASM抗碳化性能的影响机理, 以期为提高AASM抗碳化性能提供理论依据, 推进其在工程中的应用.
1 试验设计
1.1 原材料
采用巩义龙泽净水材料有限公司生产的S95级别矿渣, 其中所含CaO、 SiO2、 Al2O3、 MgO、 Fe2O3和TiO2的质量分数分别为34.29%、 35.74%、 16.45%、 9.44%、 0.52%和0.60%. 采用西陇科学股份有限公司生产的NaOH, 其纯度等级为分析纯, 其中,wNaOH≥99.0%. 采用福建品杰试验仪器有限公司生产的水玻璃原液, 水玻璃模数为3.3, 其中所含SiO2、 Na2O和H2O的质量分数分别为27.62%、 8.38%和64.00%. 试验中, 用NaOH将水玻璃的模数调至1.5. 采用河北瑞鑫矿物加工厂生产的高活性MgO, 采用辽宁洋洋高科技材料有限公司生产的中活性和低活性MgO. 根据柠檬酸法[14]测得上述高、 中、 低活性MgO的活性值分别为9.7、 124.0和393.0 s. 采用福州市闽侯县市政自来水. 采用闽江河沙作为细骨料, 其表观密度为2 568 kg·m-3、 含泥量(质量分数)为0.94%、 细度模数为2.82, 属于Ⅲ级中砂.
1.2 试验配合比
W0组(对照组)AASM中矿渣、 NaOH、 水玻璃、 水和砂的配合比(质量浓度)分别为487.7、 24.0、 187.0、 158.4和1 142.8 kg·m-3, AASM的水胶比为0.5. 在此基础上, 用活性不同的3种MgO替代AASM中质量分数为7.5 %的矿渣, 将掺有高、 中、 低活性MgO的AASM分别记为WH7.5、 WM7.5和WL7.5.
1.3 试验方法
1)宏观试验. AASM的试件制作和碳化前后抗压强度测试参照《水泥胶砂强度检验方法(GB/T 17671—2021)》[15]; AASM的吸水率测试参照《建筑砂浆基本性能试验方法标准(JCJ/T 70—2009)》[16]. 试件养护28 d, 在进行碳化试验前的2 d取出, 置于60 ℃的烘干箱内. 然后, 在试件的5个面上均匀涂抹石蜡后, 将其放入碳化箱中进行加速碳化试验. 碳化试验环境的相对湿度为(70±5)%、 温度为(20±5)℃、 CO2的体积分数为20%. 碳化深度参照《普通混凝土长期性能和耐久性能试验方法标准(GB/T 50082—2009)》[17]进行测试. 碳化后, AASM抗压强度保留率(Rc)的计算公式为
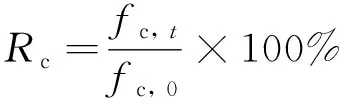
(1)
式中:fc, t为各碳化龄期AASM的抗压强度;t为碳化龄期;fc, 0为碳化前(即标准养护28 d后)AASM的抗压强度.
2)微观试验. 采用X射线粉末衍射仪(DY5261/xpert3, 美国Rigaku公司)进行AASM水化和碳化产物的X射线衍射(X-ray diffraction, XRD)分析, 扫描步长为0.02°, 扫描速度为0.01°·s-1, 2θ的测量范围为4°~90°; 采用同步热分析仪(STA449C, 德国耐驰公司)进行AASM水化和碳化产物的热重(thermal gravimetric, TG)分析, 热重分析的温度范围为室温至1 000 ℃, 加热速率为10 ℃·min-1; 采用压汞仪(AutoPore9605, 美国麦克公司)进行AASM碳化前后孔结构特征参数压汞法(mercury intrusion porosimetry, MIP)测试.
2 试验结果与讨论
2.1 吸水率和抗压强度
图1为碳化前各AASM试件的吸水率和抗压强度(fc). 由图1可知, 随着MgO活性的增加, AASM的抗压强度先上升后下降, 吸水率反之. 3种活性的MgO均可有效提高AASM的抗压强度并降低其吸水率, 与W0组相比, WL7.5、 WM7.5和WH7.5组的抗压强度分别上升2.2%、 7.1%和5.2%, 吸水率分别下降0.9%、 8.8%和3.3%. 这是由于MgO可以促进AASM的水化, 进而生成更多的类水滑石[13]; 类水滑石在一定程度上填充孔隙, 降低AASM的孔隙率, 使其变得更加密实, 从而提高其抗压强度. MgO的活性越高, AASM中生成的类水滑石越多. 当掺入质量分数为7.5%的高活性MgO时, AASM中生成过量的类水滑石, 这可能会导致AASM中出现微裂纹[18], 其密实性和抗压强度因而有所降低.

图1 AASM的吸水率和碳化前抗压强度Fig.1 Water absorption and compressive strength before carbonization of AASM
2.2 碳化深度
图2为掺入不同活性MgO后, AASM在不同碳化龄期的碳化深度. 在各组砂浆试件碳化前, 酚酞所测的碳化深度为0 mm. 由图2可知, 不同活性的MgO均可降低AASM的碳化深度. 随着MgO的活性增加, AASM的碳化深度减小. 碳化56 d后的试件WL7.5、 WM7.5、 WH7.5, 其碳化深度分别为16.53、 13.30和12.63 mm, 与试件W0相比, 其碳化深度分别降低20.9%、 36.4%和39.6%.

图2 AASM的碳化深度Fig.2 Carbonation depth of AASM
2.3 碳化后的抗压强度保留率
图3为含不同活性MgO的AASM加速碳化后的抗压强度保留率. 由图3可知, 随着碳化龄期的增加, 各AASM试件的抗压强度保留率均先降低后上升, 但始终小于100%. 这说明加速碳化后的AASM, 其强度会下降. 试件WH7.5、 WM7.5、 WL7.5和W0在各碳化龄期下的抗压强度保留率依次减小. 加速碳化56 d后, 试件WH7.5、 WM7.5、 WL7.5和W0的抗压强度保留率分别为91%、 81%、 80%和65%. 由此可见, 3种活性MgO均可减小碳化后AASM抗压强度的损失, 且MgO活性越高, AASM的抗压强度损失越小.
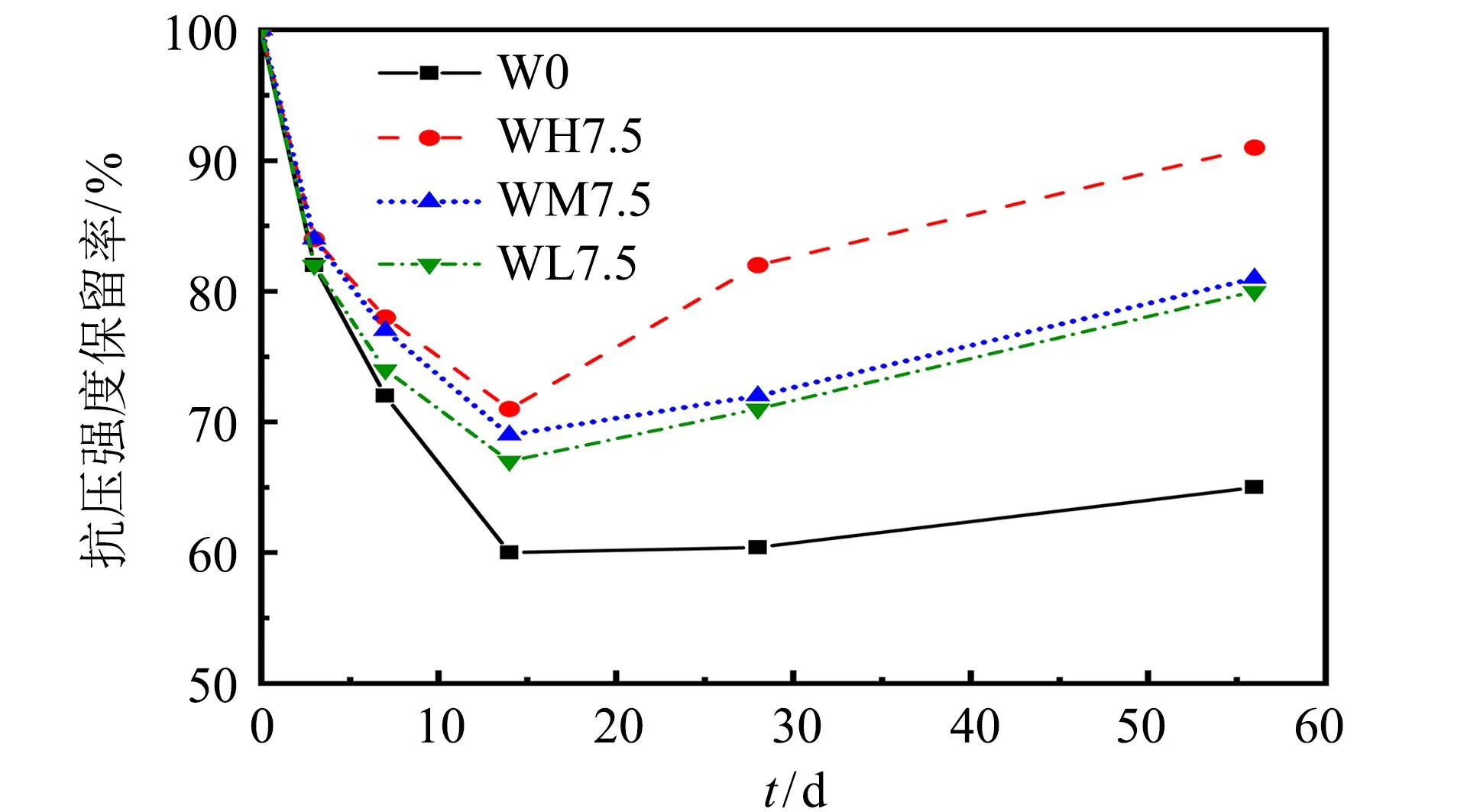
图3 碳化后AASM的抗压强度保留率Fig.3 Strength retention rate of AASM after carbonization
2.4 XRD分析
图4为加速碳化前后AASM的XRD图谱.

图4 加速碳化前后AASM的XRD图谱Fig.4 XRD patterns of AASM before carbonization and accelerated carbonization
如图4(a)所示, 碳化前, 掺入和未掺入MgO的AASM中所含主要水化产物均为水化硅(铝)酸钙(C-(A)-S-H)、 类水滑石, 还有少量的镁黄长石、 钙长石、 钙铝黄长石、 文石和方解石. 其中, 文石和方解石的出现可能是由于在制样和测试过程中试件遭受到轻微的自然碳化[19]. 此外, 掺入不同活性MgO的AASM中, 还含有镁质硅酸盐的水化物(即海泡石), 这与MgO掺入引起的Mg2+浓度增加有关. 与试件W0相比, 掺入MgO的试件中类水滑石的衍射峰更加明显, 且随着MgO活性的增加, 类水滑石的衍射峰强度增强. 另外, 在所有AASM试件中均可观察到方镁石(即MgO)的衍射峰, 而在掺入3种活性MgO的试件中, 该衍射峰更加明显. 与试件WM7.5和WL7.5相比, 试件WH7.5中方镁石的衍射峰强度更低, 说明碳化前MgO的活性越高, MgO在AASM中的反应越充分.
如图4(b)所示, 加速碳化后, AASM中生成的水化产物出现较大的变化. 与碳化前相比, 碳化后C-(A)-S-H凝胶和类水滑石的衍射峰降低, MgO的衍射峰随着MgO活性的增加而逐渐变小. 在试件WH7.5的XRD图谱中, MgO的衍射峰消失, 方解石和文石的衍射峰明显增强. 同时, 在碳化后的XRD图谱中, 还在AASM中发现新的水化产物球霰石, 且在试件WH7.5和WM7.5的图谱中还观察到水碳镁石的衍射峰. 方解石、 文石、 球霰石为3种不同晶型的CaCO3, 均为C-(A)-S-H凝胶在碳化环境下脱钙分解的产物[18]. 此外, 在碳化后的AASM中, 还发现镁方解石和菱镁矿, 分别为类水滑石和MgO与CO2之间发生碳化的产物[20]. 这说明AASM中水化产生的C-(A)-S-H凝胶和类水滑石均可与CO2发生反应, 且未反应完全的剩余MgO也可以与CO2发生反应.
2.5 TG-DTG分析
图5为加速碳化前后AASM的TG-DTG曲线.
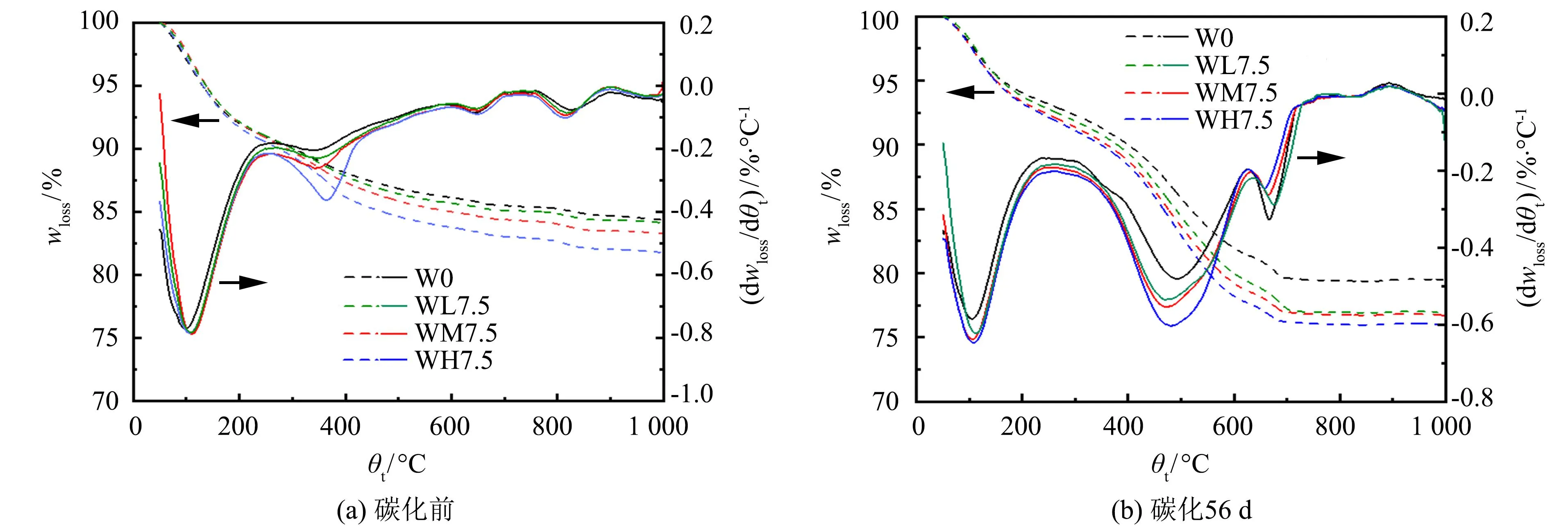
图5 加速碳化前后AASM的TG-DTG曲线Fig.5 TG-DTG curves of AASM before and after accelerated
如图5(a)所示, 在50~250、 250~400、 400~600和600~800 ℃这4个温度范围内出现较明显的分解峰. 但是, 400~600和600~800 ℃两个区间内的分解峰较小, 这是由于碳化前AASM中的水化产物未被大面积碳化. 随着温度增加, 试件质量不断降低, 这种现象在50~600 ℃区间最为明显. 从DTG曲线可以看出, 在50~250和250~400 ℃区间出现两个明显的分解峰, 分别对应C-(A)-S-H凝胶层间水的蒸发和类水滑石的脱水[19-20]. 通过比较这两个温度范围的质量损失和分解峰的大小可知, 在掺入MgO的试件中, C-(A)-S-H凝胶和类水滑石的质量损失均增大, 说明3种不同活性的MgO均具有促进C-(A)-S-H凝胶和类水滑石生成的作用. 其中, 试件WH7.5中类水滑石对应的分解峰要明显高于试件WM7.5和WL7.5, 说明高活性的MgO更能促进AASM的水化, 生成更多类水滑石. 试件WM7.5、 WH7.5、 WL7.5、 W0中C-(A)-S-H凝胶的分解峰强度依次减小, 这与碳化前各试件抗压强度的大小关系一致(见图1).
如图5(b)所示, 碳化后在50~250、 250~400、 400~600和600~800 ℃这4个温度范围内也出现较明显的分解峰. 但是, 与碳化前相比, 50~250 ℃区间内C-(A)-S-H凝胶失水所对应的分解峰减弱. 说明在加速碳化环境下, AASM中的C-(A)-S-H凝胶脱钙分解, 导致凝胶减少. 因此, 碳化后AASM的抗压强度保留率变小(见图3). 400~600和600~800 ℃两个区间内的分解峰明显增强. 400~600 ℃区间内的分解峰是因类水滑石的脱碳、 碳化产物水碳镁石的失水、 菱镁矿的分解等分解峰叠加而形成的. 600~800 ℃区间内的分解峰是因碳化产物方解石/镁方解石的分解而形成的[21-22].
由图5(b)还可以看出, 掺入3种活性不同的MgO后, 随着MgO活性的增加, C-(A)-S-H凝胶失水所对应的分解峰明显增强, 而方解石/镁方解石分解所对应的分解峰明显减弱. 这说明在加速碳化后, 掺有MgO的试件中含有更多的类水滑石和未反应的MgO, 它们会与CO2反应, 减少C-(A)-S-H凝胶碳化脱钙分解生成的方解石/镁方解石, 保留更多的C-(A)-S-H凝胶. 此外, MgO活性越高, AASM中保留的C-(A)-S-H凝胶越多. 因此, 碳化后AASM的抗压强度保留率随着MgO活性的增加而增加(见图3).
2.6 孔结构分析
表1和图6分别为AASM在加速碳化前后的孔隙结构特征参数和孔径(d)分布曲线. 表1中:φ总为总孔隙率;k为曲折度.
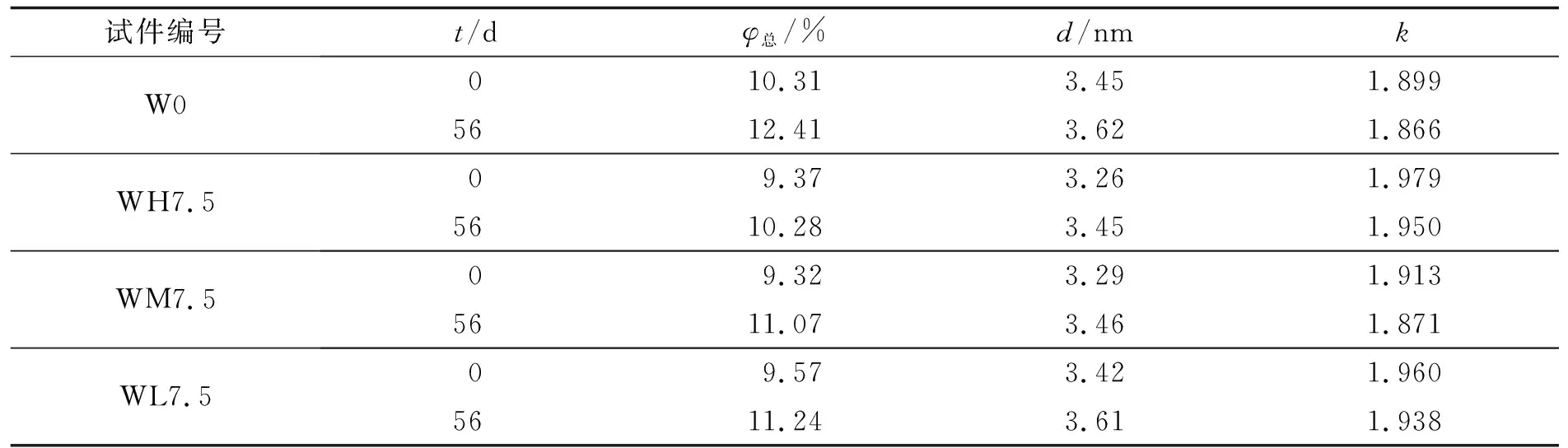
表1 加速碳化前后AASM的孔隙结构特征参数Tab.1 Pore structure characteristic parameters of AASM before and after accelerated carbonization

图6 加速碳化前后AASM的孔径分布Fig.6 Pore size distribution of AASM before and after accelerated carbonization
由表1可知, 掺入3种不同活性MgO后的AASM, 其总孔隙率和最可几孔径均低于试件W0, 孔隙的曲折度均高于试件W0. 随着MgO活性的增加, AASM的最可几孔径和总孔隙率先降低后增加, 这可以解释图1中吸水率和抗压强度的变化规律. 由图6(a)可知, 加速碳化前, AASM的孔径主要分布在3~10和1 000~10 000 nm这两个区间. 在3~10 nm区间内, 与试件W0相比, 试件WL7.5的峰值略有降低, 试件WM7.5和WH7.5的峰值均有所升高, 且试件WH7.5的孔径分布曲线由试件W0的一个峰转变为两个峰. 在1 000~10 000 nm区间内, 3种活性的MgO均可使峰值降低. 随着MgO活性的增加, 峰值先降低后增加, 且始终低于试件W0的峰值. 这说明MgO的掺入可以优化AASM的孔结构. MgO的掺入可促进AASM的水化, 使其生成更多的水化产物, 从而填充内部空隙. 但是, 高活性的MgO会使AASM的水化更加剧烈, 产生较多的类水滑石, 导致微裂纹的产生[23]. 因此, 试件WH7.5的孔结构比试件WM7.5差.
由表1还可知, 与碳化前的AASM相比, 碳化后4组AASM的总孔隙率和最可几孔径均增大, 孔隙的曲折度均减小, 这说明碳化会使AASM的孔结构劣化. 但是, 掺入MgO的试件, 其总孔隙率和最可几孔径均小于试件W0, 孔隙曲折度均大于试件W0, 说明掺入MgO可以缓解孔结构的劣化. 随着MgO活性的增加, AASM的总孔隙率和最可几孔径逐渐变小, 说明MgO活性越大, 对AASM孔结构的改善效果越好. 由图6(b)可知, 各组试件的孔径主要分布在3~10和1 000~10 000 nm这两个区间. 与加速碳化前的AASM相比, 碳化后AASM在3~10 nm区间内的峰有较明显的减弱. 在加速碳化后, 试件WH7.5在3~10 nm区间内的峰由两个峰转变为一个峰, 并且该峰的峰值较其它试件更高. 从孔径大于200 nm开始, 各试件的峰值升高, 在1 000~10 000 nm区间内的峰显著降低, 并且随着MgO活性的增加, 各试件的峰值逐渐降低. 这说明MgO的掺入可以减小AASM的孔隙率, 抑制CO2在AASM中扩散, 减小AASM的碳化深度, 提高其抗碳化性能. 此外, 掺入MgO的活性越大, AASM的抗碳化性能越好.
3 结语
1) MgO的掺入能促进AASM的水化, 产生C-(A)-S-H凝胶和类水滑石. MgO的活性越大, AASM水化产生的类水滑石越多.
2) 与未掺入MgO的AASM相比, 掺入不同活性MgO的AASM, 其密实性和抗压强度均有所提高. MgO的活性越高, AASM的水化越剧烈. 过多类水滑石的产生会引起AASM中微裂纹的产生和孔结构的劣化. 因此, 试件WM7.5的密实性和抗压强度略高于试件WH7.5.
3) 加速碳化使AASM中的C-(A)-S-H凝胶脱钙分解, 其总孔隙率增加, 抗压强度下降. 掺入MgO 后, AASM中产生更多的类水滑石, 未反应的MgO会与CO2反应, 进而降低C-(A)-S-H凝胶的分解程度和总孔隙率的增加程度. AASM的碳化深度因此降低, 抗压强度保留率提高.
4) MgO活性越高, AASM中就有越多的类水滑石与CO2发生反应. 更多的CO2被消耗, 会引起C-(A)-S-H凝胶碳化脱钙分解程度和总孔隙率增加程度降低. AASM的碳化深度因此降低, 抗压强度保留率提高.
猜你喜欢
硅酸盐通报(2022年8期)2022-09-08 04:25:42
矿产综合利用(2021年3期)2021-07-14 03:46:54
矿产综合利用(2020年1期)2020-07-24 08:50:46
中国环境监察(2017年8期)2017-10-23 05:25:14
材料科学与工程学报(2016年4期)2017-01-15 13:35:40
中国塑料(2016年4期)2016-06-27 06:33:28
中国塑料(2016年11期)2016-04-16 05:25:56
中国塑料(2015年6期)2015-11-13 03:03:11
中国塑料(2015年5期)2015-10-14 00:59:49
化学反应工程与工艺(2015年3期)2015-04-16 03:06:20
