
整合生物信息学分析AURKA 基因在宫颈癌中的表达及临床意义
2023-12-23 13:43:00王秋李晓峰夏勇熊丹
分子诊断与治疗杂志 2023年11期
王秋 李晓峰 夏勇 熊丹
宫颈癌是全球范围内最常见的女性恶性肿瘤之一,死亡率在女性恶性肿瘤中居第二位[1-2]。85%的宫颈癌死亡发生在中低等收入人群国家[3]。目前,宫颈癌的筛查、手术、放化疗等治疗方法已经取得一定的成效,但是对晚期和复发性宫颈癌的临床治疗效果还远远不够,患者的预后较差[4]。因此,迫切需要更好的宫颈癌治疗靶点和检测标志物来提高患者的生存期。
当前,在预防癌症方面有许多公共数据库可供研究。重新分析与宫颈癌相关的基因表达谱对于其发病机制和分子机理非常重要。许多研究已探讨了宫颈癌的致病基因及预后[5],如研究发现INHBA(TGF-β 超家族的成员之一)在宫颈癌中高度表达,并与宫颈癌的不良预后相关。同时,INHBA 也与免疫细胞浸润相关[6]。Xue 等[7]研究发现MCM2可能与宫颈癌的发生和发展有关,并进一步验证了MCM2的敲除可以抑制宫颈癌细胞的增殖。然而,基因之间以各种方式相互作用,肿瘤基因组复杂多变,现行的研究结果远不足以揭示宫颈癌的发病机制及预后预测。因此,需要从不同角度进行研究和分析,本研究拟使用生物信息学方法通过多组学分析寻找宫颈癌的新型预后生物标志物。
本研究确定了GSE9750、GSE46857和GSE52903数据库中的差异表达基因(Differentially Expressed Genes,DEGs),对这些差异表达基因进行了GO、KEGG 和蛋白互作网络等多种方法分析并得出枢纽基因。进一步对枢纽基因的临床意义(包括表达和生存分析)进行分析,为进一步了解这些枢纽基因在宫颈癌的发展和转移中的作用提供新的方向,同时加深对宫颈癌发生发展的分子机制的认识,为宫颈癌的治疗提供新的潜在靶点。
1 材料和方法
1.1 材料来源
GEO 1据库(https://www.ncbi.nlm.nih.gov/gds)用于下载微阵列数据集。分别下载GSE9750、GSE46857 和GSE52903 数据集进行分析。研究过程见图1。AURKA基因免疫组化用的抗体来源于Abcam(货号:ab52973)。
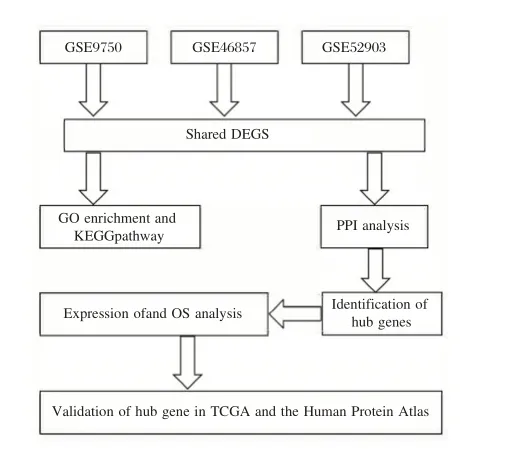
图1 研究路线图Figure 1 The study procedure
1.2 基因富集和功能分析
使用在线程序仙桃学术(https://www.xiantao.love/)对基因功能进行注释(GO 分析)和KEGG 通路进行富集分析。
1.3 差异表达基因(DEGs)的选择
使 用GEO2R 工 具(http://www.ncbi.nlm.nih.gov/geo/geo2r/)得到DEGs,并设置|log2fold change|≥1 和P<0.05 为临界值。
1.4 PPI 网络和模块分析
筛选得到上调和下调的DEGs,使用String 数据库(http://string-db.org)进行分析,设置临界值大于0.4。使用Cytoscape v3.9.0 插件从P<0.01 中选择PPI 网络(https://cytoscape.org/)的显著模块,并由MCODE 插件进一步分析。
1.5 生存分析
通过使用Kaplan-Meier plotter(KM plotter,http://www.kmplot.com)对枢纽基因进行总生存期和无复发生存期分析。
1.6 宫颈癌枢纽基因表达的验证
GEPIA 数据库是基于TCGA 创建的数据库,在该数据库上进行枢纽基因mRNA 表达验证。枢纽基因基因的蛋白表达在人类蛋白图谱(http://www.proteinatlas.org/)中进行验证。
1.7 免疫组化
标本经10% 中性福尔马林固定、常规脱水、浸蜡、石蜡包埋,4 μm 厚连续切片,免疫组织化学染色使用罗氏Bench-Mark ULTRA 全自动免疫组织化学染色机进行染色,一抗AURKA 试剂购自Abcam 公司(ab52973)。
1.8 统计学方法
采用R 4.2.2 版本软件(美国Math-Soft 公司)进行统计和绘图。研究连续变量通过t检验进行分析比较。生存分析采用Kaplan-Meier 统计学方法进行分析。以P<0.05 为差异有统计学意义。
2 结果
2.1 差异表达基因(DEGs)的筛选和分析
使用火山图显示DEGs,红色点代表上调的DEGs,蓝色点代表下调的DEGs。见图2A~2C。在GSE9750、GSE46857 和GSE52903 中分别鉴定出上调基因610,747,355 个,下调基因1 572,700,502 个。利用维恩图共鉴定出30 个共同上调的DEGs 和49 个共同下调的DEGs。见图2D~2E。

图2 不同数据集中的差异表达基因Figure 2 Differentially expressed genes in the different datasets
2.2 GO 和KEGG 富集分析
对图2 鉴定出来的30 个上调的DEGs 和49 个下调的DEGs 进行GO 和KEGG 富集分析。GO 分析:“生物过程”前三位为“细胞器裂变”、“细胞器组织的负调控”、“再生”;“细胞组分”前三位为“含胶原蛋白的细胞外基质”、“核染色体”、“细胞周期蛋白依赖性蛋白激酶全酶复合物”;“分子功能”前三位为“蛋白质C 端结合”、“整合素结合”、“细胞外基质结合”。KEGG 分析DEGs 富集的通路主要为“细胞周期”、“孕酮介导的卵细胞成熟”和“铂耐药性”。GO 分析和KEGG 分析结果显示采用气泡图。见图3A。或采用网络图。见图3B。
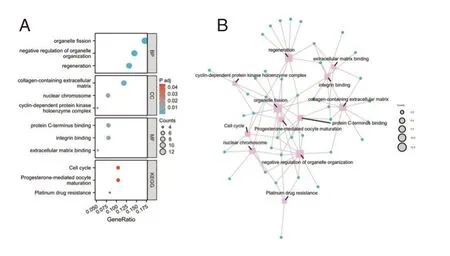
图3 差异表达基因的GO 和KEGG 分析的结果Figure 3 Results of the GO and KEGG analyses of the differentially expressed genes
2.3 蛋白质相互作用网络及枢纽基因生存分析
构建蛋白质相互作用网络。见图4A。从PPI网络中识别出得分最高的前15 个基因。见图4B。通过KM plotter 分析出15 个枢纽基因在宫颈癌总生存期(OS)和无复发生存期(RFS)中的作用,在OS 分析中包含了304 例宫颈癌患者,发现AURKA基因的高表达与宫颈癌患者总生存期较短显著相关。见图5A。RFS 分析中包含了174 例宫颈癌患者,发现AURKA基因的高表达与宫颈癌患者无复发生存期较短显著相关。见图5B。

图4 差异表达基因的PPI 网络和模块分析Figure 4 PPI network and module analysis of DEGs
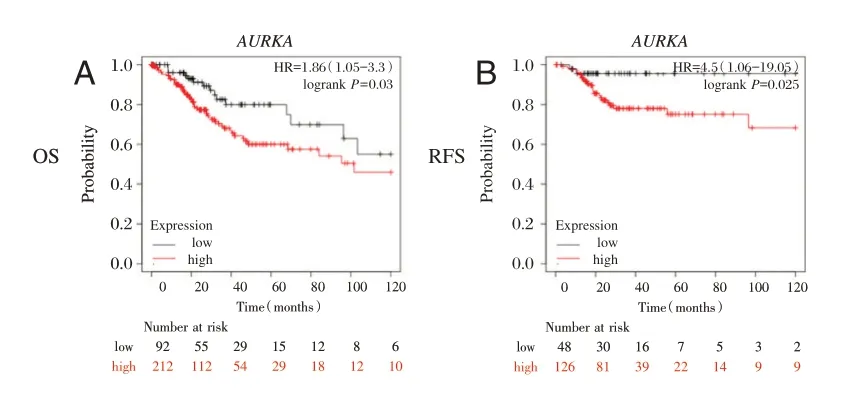
图5 枢纽基因的OS 及RFS 的相关性分析Figure 5 The correlation analysis between hub genes and OS or RFS of cervical carcinoma patients
2.4 枢纽基因在宫颈癌中表达的验证及临床应用价值分析
AURKA基因在宫颈癌组织中上调。见图6A。在不同种族人群中的表达差异无统计学意义(P>0.05)。见图6B。但在人类蛋白图谱中,AURKA基因在宫颈癌组织的蛋白表达水平显著高于正常宫颈组织图。见图6C。采用受试者工作特征(ROC)曲线来评估AURKA基因表达水平对区分宫颈癌组织和正常宫颈组织的预测价值。见图6D。ROC 曲线下面积(AUC)为0.999。收集10 对临床样本进行AURKA基因在宫颈癌及癌旁组织中表达的免疫组化验证,结果显示,AURKA基因在宫颈癌组织中表达增高。见图7A~7B。差异有统计学意义(P<0.05)。见图7C。不同分化程度癌组织表达差异无统计学意义(P>0.05)。见图7D-7F。

图6 AURKA 基因在宫颈癌中表达的验证Figure 6 Validation of the expression of the AURKA genes in cervical carcinoma

图7 AURKA 基因在宫颈癌组织中的表达Figure 7 The expression of the AURKA genes in cervical carcinoma tissues
3 讨论
宫颈癌是起源于子宫颈的恶性肿瘤,是女性生殖系统中最广泛的恶性肿瘤[8-9]。宫颈癌的发生发展过程与多种遗传和环境因素相关[10]。为了识别潜在的生物标志物,仍需要进行大量详细的研究,包括治疗方案、途径、机制和预后。本研究中,分析了来自三个GEO 数据集(GSE9750、GSE46857 和GSE52903)的差异表达基因,得到30 个基因表达上调,49 个基因表达下调,对差异基因进行GO 和KEGG 通路研究发现差异表达基因主要与“细胞周期”、“孕酮介导的卵细胞成熟”和“铂耐药性”信号通路有关。此外,通过分子相互作用的分析,进一步筛选出前15 个枢纽基因并进行了预后生存分析,发现了AURKA基因的高表达与宫颈癌患者总生存期较短显著相关。同样,RFS 分析发现AURKA基因的高表达与宫颈癌患者无复发生存期较短相关。对AURKA基因的表达进行验证发现其在宫颈癌组织中mRNA 和蛋白的表达均上调,但无种族人群差异。最后,通过ROC 曲线分析进一步表明AURKA基因在宫颈癌中具有较好的预测价值。
AURKA是一种丝氨酸/苏氨酸激酶,在细胞有丝分裂中起着重要的调节作用[11]。AURKA在许多恶性肿瘤中高表达,是中心体复制、成熟和分离的关键[12]。这与本研究GO 和KEGG 分析得到的结果吻合。研究表明,AURKA在大多数人类癌症中升高与肺癌、肝癌和结直肠癌患者的不良预后密切相关[13-15]。AURKA作为一种致癌基因,影响许多肿瘤的生物学过程,如增殖、基因组的不稳定性、侵袭和转移[16]。Liu 等[17]研究发现,AURKA抑制剂可通过与AURKA结合诱导G2/M 期阻滞和线粒体诱导结肠癌细胞内源性凋亡。Shao 等[18]报道了子宫颈癌患者组织中AURKA的表达明显增加。进一步研究表明,hsa_circ_0075341 通过隔离miR-149-5p 和上调AURKA的表达来增强宫颈癌细胞的增殖和侵袭能力。Van Horn 等[19]研究发现,CDK1 的激活启动了AURKA所参与的有丝分裂的激活[19]。此外,被激活的AURKA和PLK1 进一步在作用于CDK1,促进有丝分裂,并参与有丝分裂中的纺锤体形成和中心粒分裂。近期,Wang 等[20]综合多个GEO 数据集分析发现CDK1 和PRC1 与良好的生存期相关,而AURKA与较差的生存期相关。本研究综合GSE9750、GSE46857 和GSE52903数据集分析,发现AURKA在宫颈癌中表达显著增高,进一步研究发现AURKA与宫颈癌的总生存期及无复发生存期密切相关,TCGA 数据库和临床样本进一步验证了AURKA在宫颈癌的高表达,且无种族人群差异。重要的是ROC 曲线分析亦显示AURKA在宫颈癌中具有较好的预测价值。
综上所述,AURKA可能作为宫颈癌检测和靶向治疗的一个新靶点。
猜你喜欢
中老年保健(2021年12期)2021-08-24 03:30:20
房地产导刊(2020年8期)2020-09-11 07:47:38
商周刊(2019年18期)2019-10-12 08:50:56
当代陕西(2018年12期)2018-08-04 05:49:06
西南国防医药(2016年6期)2016-12-01 06:01:08
河北医学(2016年5期)2016-12-01 03:58:55
癌症进展(2016年11期)2016-03-20 13:16:04
食管疾病(2015年3期)2015-12-05 01:45:07
哈尔滨医药(2015年6期)2015-12-01 03:58:17
中医研究(2014年8期)2014-03-11 20:29:19
