
三个含4,4′-(吡啶-3,5-二基)二苯甲酸配体的锌(Ⅱ)、镍(Ⅱ)和钴(Ⅱ)配位聚合物的合成、晶体结构及催化性质
2023-12-21 01:01亢秀琪王嘉浩顾金忠
无机化学学报 2023年12期
亢秀琪 王嘉浩 顾金忠
(兰州大学化学化工学院,兰州 730000)
Coordination polymers have been widely explored as an important family of compounds with a high diversity of structures[1-4], and remarkable functional properties, such as sensing[5-6], gas sorption[7-8], and catalysis[9-11]. This is also governed by some features of metal ions and derived compounds, such as low cost and abundance,bio-and photoactivity,as well as rich coordination chemistry toward a broad spectrum of ligand types[12-14]. Among them, aromatic polycarboxylic acids stand out as the most commonly applied linkers for assembling coordination polymers and derived materials[4,10-11]. A considerable focus of current research is devoted to the design of novel carboxylate linkers with multiple potential sites for coordination, followed by their exploration of the synthesis of metal-organic architectures.
In pursuit of our general research line on probing various commercially available but still rarely used polycarboxylic acids as linkers for designing functional coordination polymers[10-11,15-16], in the current work we have chosen a biphenylpyridine-dicarboxylic acid:4,4′-(pyridin-3,5-diyl)dibenzoic acid (H2pdba, Scheme 1).The organic compound can potentially act as a versatile semi-flexible linker for the synthesis of coordination polymers. The selection of the ligand has relied on the following considerations. (1) The ligand precursor features up to four O-donors and one Npyridinesite for potential coordination to metal modes. (2) The dicarboxylic acid is stable under hydrothermal synthetic conditions, semi-flexible due to the presence of C—C bonds between adjacent aromatic rings, and can exhibit different degrees of deprotonation and a variety of coordination modes. (3) H2pdba has not yet been widely applied in the field of coordination polymers[17-19].
Therefore, based on the above reasons, we designed and synthesized three Zn(Ⅱ), Ni(Ⅱ), and Co(Ⅱ)coordination polymers based on a biphenylpyridinetype dicarboxylate ligand.Herein,we report the syntheses, crystal structures, and catalytic properties of these coordination polymers constructed from the biphenylpyridine-type dicarboxylate ligand.
1 Experimental
1.1 Reagents and physical measurements
All chemicals and solvents were of AR grade and used without further purification. Carbon, hydrogen,and nitrogen were determined using an Elementar Vario EL elemental analyzer. IR spectra were recorded on a Bruker EQUINOX 55 spectrometer using KBr pellets. Thermogravimetric analysis (TGA) data were collected on a LINSEIS STA PT1600 thermal analyzer with a heating rate of 10 ℃·min-1. Powder X-ray diffraction (PXRD) patterns were measured on a Rigaku-Dmax 2400 diffractometer using CuKαradiation (λ=0.154 06 nm); the X-ray tube was operated at 40 kV and 40 mA; the data collection range was between 5°and 45°. Solution1H NMR spectra were recorded on a JNM ECS 400M spectrometer.
1.2 Synthesis of{[Zn(μ3-pdba)(phen)]·H2O}n(1)
A mixture of ZnCl2(0.027 g, 0.20 mmol), H2pdba(0.064 g, 0.20 mmol), phen (0.040 g, 0.20 mmol),NaOH (0.016 g, 0.40 mmol), and H2O (10 mL) was stirred at room temperature for 15 min,and then sealed in a 25 mL Teflon-lined stainless steel vessel,and heated at 160 ℃for 3 d, followed by cooling to room temperature at a rate of 10 ℃·h-1.Colourless block-shaped crystals of 1 were isolated manually, and washed with distilled water. Yield: 51 % (based on H2pdba). Anal.Calcd. for C31H21ZnN3O5(%): C 64.09, H 3.64, N 7.23;Found(%): C 64.42, H 3.62, N 7.20. IR (KBr, cm-1):3 167w,3 064w,2 924w,1 601s,1 544m,1 528w,1 397s,1 328w,1 237w,1 193w,1 105w,1 014w,977w,909w,869w,838w,786m,725w,710w,681w,649w.
1.3 Synthesis of{[Ni(μ3-pdba)(bipy)]·3H2O}n(2)
A mixture of NiCl2·6H2O (0.048 g, 0.20 mmol),H2pdba (0.064 g, 0.20 mmol), bipy (0.031 g, 0.20 mmol), NaOH (0.016 g, 0.40 mmol), and H2O (10 mL)was stirred at room temperature for 15 min,then sealed in a 25 mL Teflon-lined stainless steel vessel,and heated at 160 ℃for 3 d, followed by cooling to room temperature at a rate of 10 ℃·h-1. Green block-shaped crystals of 2 were isolated manually, washed with distilled water, and dried. Yield: 54% (based on H2pdba).Anal. Calcd for C29H25NiN3O7(%): C 59.42, H 4.30, N 7.17; Found(%): C 59.16, H 4.27, N 7.20. IR (KBr,cm-1):3 463w,3 072w,2 364w,1 601s,1 552m,1 413s,1 324w,1 237w,1 188w,1 165w,1 109w,1 017w,938w,909w,862w,814w,781m,737w,710w,674w.
1.4 Synthesis of {[Co(μ3-pdba)(H2biim)(H2O)]·H2O}n(3)
A mixture of CoCl2·6H2O (0.048 g, 0.2 mmol),H2pdba (0.064 g, 0.20 mmol), H2biim (0.027 g, 0.2 mmol), NaOH (0.016 g, 0.4 mmol), and H2O (10 mL)was stirred at room temperature for 15 min, and then sealed in a 25 mL Teflon-lined stainless steel vessel,and heated at 160 ℃for 3 d, followed by cooling to room temperature at a rate of 10 ℃·h-1. Orange blockshaped crystals of 3 were isolated manually, and washed with distilled water. Yield: 47% (based on H2pdba).Anal.Calcd.for C25H21CoN5O6(%):C 54.95,H 3.87,N 12.82;Found(%):C 55.26,H 3.85,N 12.71.IR(KBr, cm-1): 3 224w, 3 124w, 2 796w, 1 604s, 1 552m,1 525w,1 433w,1 369s,1 253w,1 173w,1 105w,1 014w,902w,789m,750w,705w,661w.
These compounds are insoluble in water and common organic solvents, such as methanol, ethanol,acetone,and DMF.
1.5 Structure determination
The data of three single crystals with dimensions of 0.12 mm×0.09 mm×0.08 mm (1), 0.11 mm×0.10 mm×0.08 mm (2),and 0.11 mm×0.06 mm×0.05 mm (3)were collected at 293(2) K on a Bruker SMART APEXⅡCCD diffractometer with CuKα(λ=0.154 184 nm).The structures were solved by direct methods and refined by full-matrix least-square onF2using the SHELXTL-2014 program[20]. All non-hydrogen atoms were refined anisotropically. All the hydrogen atoms(except those bound to water molecules) were placed in calculated positions with fixed isotropic thermal parameters included in structure factor calculations in the final stage of full-matrix least-squares refinement. The hydrogen atoms of water molecules in 3 were located by different maps and constrained to ride on their parent O atoms. Some highly disordered solvent molecules in 1 and 2 were removed using the SQUEEZE routine in PLATON[21]. The final amount of solvent molecules was estimated from the data of elemental and thermogravimetric analyses. A summary of the crystallography data and structure refinements for 1-3 is given in Table S1 (Supporting information). The selected bond lengths and angles for compounds 1-3 are listed in Table S2.Hydrogen bond parameters of compound 3 are given in Table S3.
CCDC:2271015,1;2271016,2;2271017,3.
2 Results and discussion
2.1 Description of the structure
2.1.1 Crystal structure of compound 1
Single-crystal X-ray diffraction analysis reveals that compound 1 crystallizes in the monoclinic space groupP21/c. The asymmetric unit of 1 contains one crystallographically unique Zn (Ⅱ)ion, oneμ3-pdba2-block, one phen moiety, and one lattice water molecule.As shown in Fig.1,Zn1 ion is five-coordinated by two carboxylate O and one N atoms from three individualμ3-pdba2-blocks and two N atoms from the phen ligand, constructing a distorted trigonal bipyramidal[ZnN3O2] geometry. The Zn—O bond lengths range from 0.196 9(3) to 0.203 0(2) nm, whereas the Zn—N bonds vary from 0.214 9(3) to 0.221 1(3) nm; these bonding parameters are comparable to those found in other reported Zn(Ⅱ)compounds[4,15-16]. In 1, the pdba2-ligand adopts coordination mode Ⅰ(Scheme 1), in which two deprotonated carboxylate groups show a monodentate mode. Theμ3-pdba2-blocks connect adjacent Zn(Ⅱ)ions to form a 2D network (Fig.2). From a topological viewpoint, it is composed of the 3-linked Zn1 andμ3-pdba2-nodes (topologically equivalent) that are arranged into a mononodal 3-connected net with ahcb(Shubnikov hexagonal plane net/(6,3))topology and point symbol of (63) (Fig.3).A significant feature of this structure also concerns its two-fold parallel 2D+2D interpenetration(Fig.4).
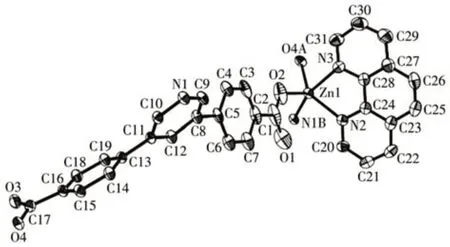
Fig.1 Drawing of the asymmetric unit of compound 1 with a 30% probability of thermal ellipsoids
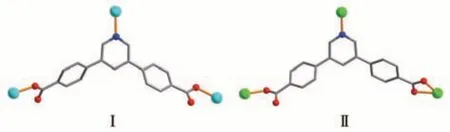
Scheme 1 Coordination modes of pdba2-ligands in compounds 1-3
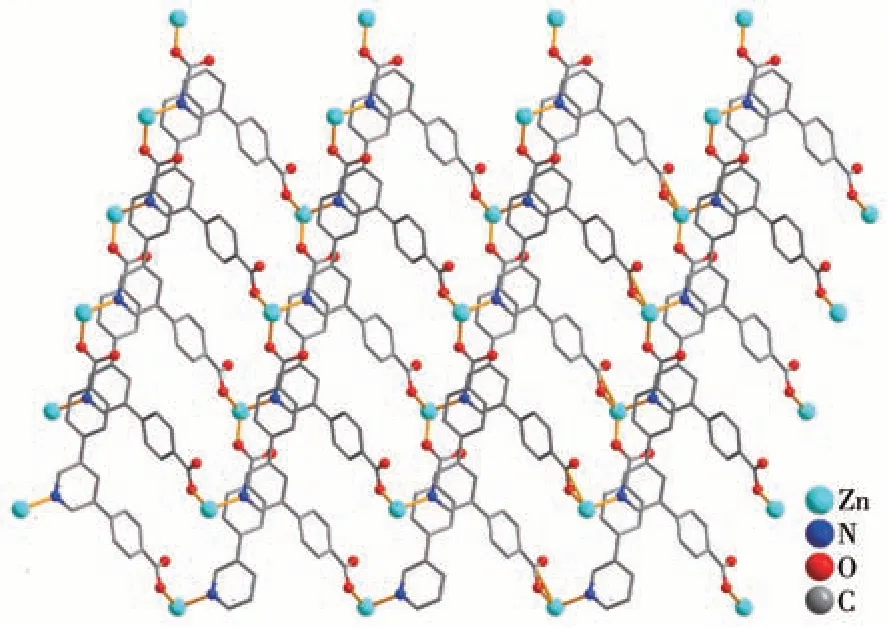
Fig.2 Two-dimensional metal-organic network viewed along the b-axis
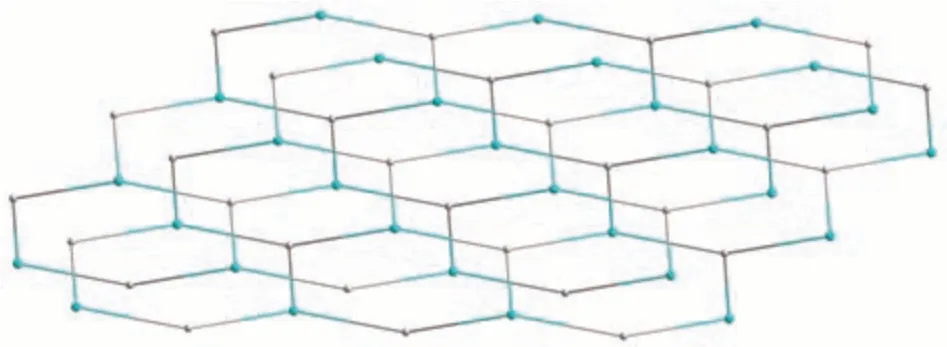
Fig.3 Topological representation of an uninodal 3-connected metal-organic layer with an hcb topology viewed along the b-axis
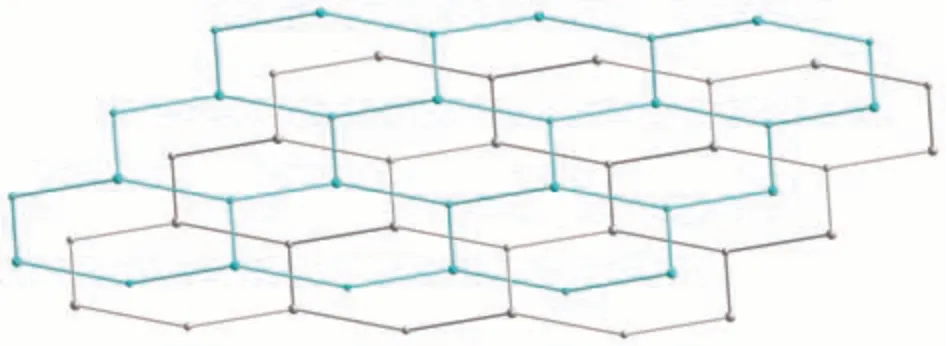
Fig.4 Two 2D+2D interpenetrated hcb layers shown by different colors(cyan and gray)
2.1.2 Crystal structure of compound 2
The asymmetric unit of 2 possesses one crystallographically independent Ni(Ⅱ)ion,oneμ3-pdba2-block,one bipy ligand, and three lattice water molecules. As shown in Fig.5, the Ni1 ion is six-coordinated and adopts a distorted octahedral [NiN3O3] geometry formed by three carboxylate O and one N atoms from three distinctμ3-pdba2-blocks and a pair of Nbipyatoms.The Ni—O distances range from 0.201 51(18) to 0.214 74(19) nm, whereas the Ni—N distances are 0.207 7(2)-0.209 2(2) nm; these bonding parameters agree with those observed in other Ni (Ⅱ) compounds[15,22]. In 2, the pdba2-block also acts as aμ3-spacer (mode Ⅱ, Scheme 1), in which the carboxylate groups exhibit the monodentate or bidentate modes.The neighboring Ni (Ⅱ)ions are linked byμ3-pdba2-blocks, giving rise to a 2D sheet (Fig. 6). This 2D network also discloses a mononodal 3-connected framework with anhcbtopology (Fig.7)and a point symbol of(63).A significant feature of this structure also concerns its two-fold parallel 2D+2D interpenetration(Fig.8).
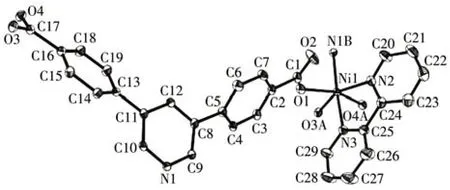
Fig.5 Drawing of the asymmetric unit of compound 2 with a 30% probability of thermal ellipsoids

Fig.6 Two-dimensional metal-organic network viewed along the b-axis
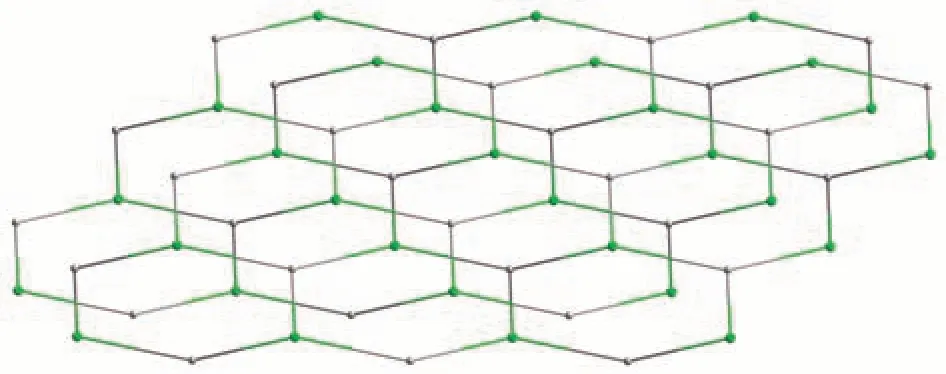
Fig.7 Topological representation of an uninodal 3-connected metal-organic layer with an hcb topology viewed along the b-axis
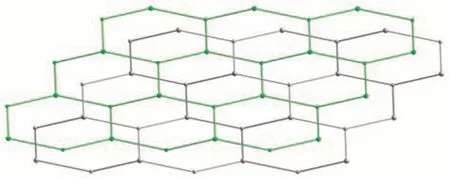
Fig.8 Two 2D+2D interpenetrated hcb layers shown by different colors(green and gray)
2.1.3 Crystal structure of compound 3
This compound has a 1D metal-organic chain structure. An asymmetric unit contains a Co(Ⅱ)center,aμ5-pdba2-block, an H2biim moiety, an H2O ligand,and a lattice water molecule. The Co1 center is sixcoordinated and assumes a distorted octahedral[CoN3O3] environment, which is populated by two carboxylate O atoms from twoμ3-pdba2-blocks,one H2O ligand, and two N donors of the H2biim moiety (Fig.9).The Co—O (0.206 4(2)-0.216 8(2) nm) and Cd—N(0.214 8(3)-0.217 9(3) nm) bonds show typical distances[10-11]. The pdba2-block acts as aμ3-spacer (mode Ⅰ,Scheme 1), in which both COO-groups are monodentate. The adjacent Co (Ⅱ)ions are connected viaμ3-pdba2-blocks to form a 1D metal-organic chain(Fig.10). The 1D ladder-like chain in this structure is defined as a mononodal 3-connected net(Fig.11)with a(4,4)(0,2)topology and a point symbol of(42.6).
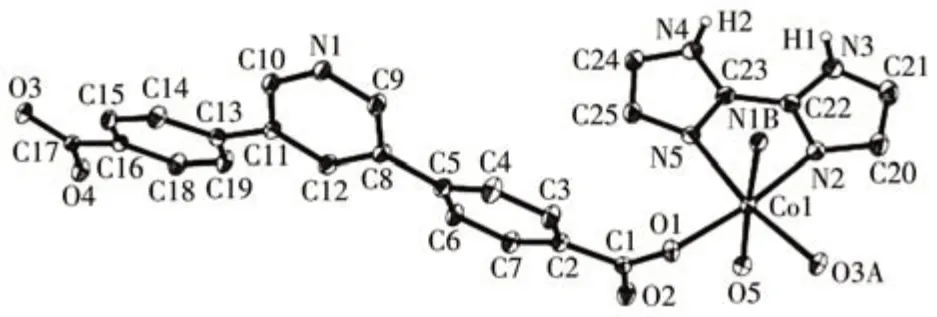
Fig.9 Drawing of the asymmetric unit of compound 3 with a 30% probability of thermal ellipsoids
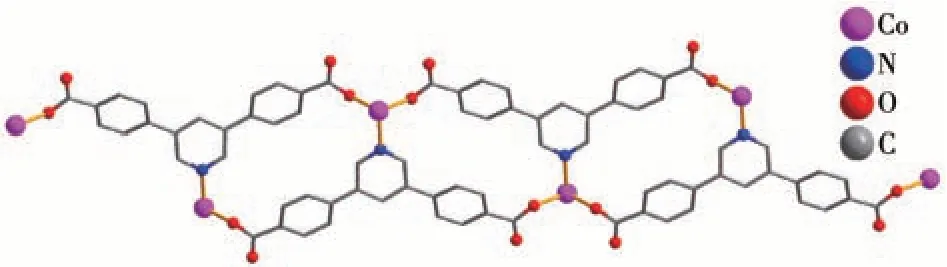
Fig.10 One-dimensional metal-organic chain viewed along the b-axis
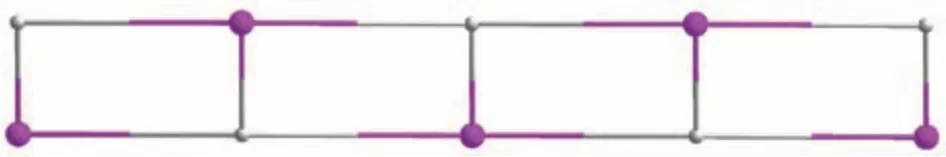
Fig.11 Topological representation of a mononodal 3-connected double chain with a(4,4)(0,2)topology viewed along the b-axis
2.2 Thermal stability of the compounds
To determine the thermal stability of polymers 1-3, their thermal behaviors were investigated under a nitrogen atmosphere by TGA.As shown in Fig.12,compound 1 lost its one lattice water molecule in a range of 79-111 ℃(Obsd. 3.2%, Calcd. 3.1%), followed by the decomposition at 312 ℃. In 2, there was a release of three lattice water molecules between 34 and 176 ℃(Obsd.9.4%,Calcd.9.2%),whereas a dehydrated sample remained stable up to 313 ℃.For 3,the TGA curve displayed a loss of two water molecules (one lattice and one coordinated)between 173 and 252 ℃(Obsd.6.4%,Calcd. 6.6%), whereas a dehydrated solid was then stable up to 305 ℃.
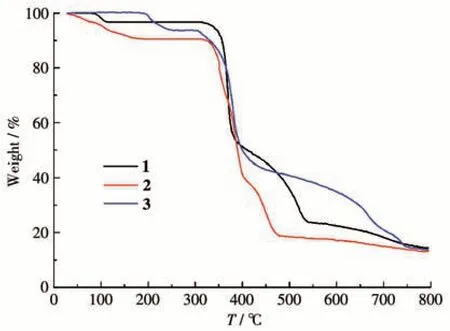
Fig.12 TGA curves of compounds 1-3
2.3 Catalytic Henry reaction
Considering the potential of different metal (Ⅱ)coordination compounds to act as catalysts in the Henry condensation[16,23-24], we probed the compounds 1-3 as heterogeneous catalysts in this reaction using assorted aldehydes with nitromethane. As a model substrate, 4-nitrobenzaldehyde was treated with nitromethane at 70 ℃in methanol medium to form a 2-nitro-l-(4-nitrophenyl)ethan-1-ol product (Scheme 2, Table 1).The influence of different reaction parameters (i.e.,reaction time, temperature, solvent type, catalyst loading and recycling,and substrate scope)was studied.
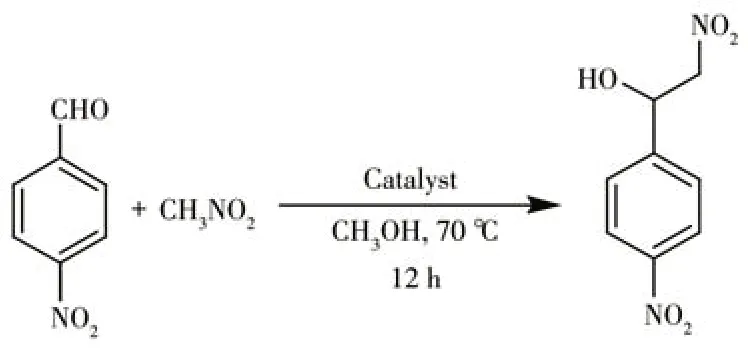
Scheme 2 Henry reaction of 4-nitrobenzaldehyde with nitromethane(model substrate)catalyzed by the coordination polymers
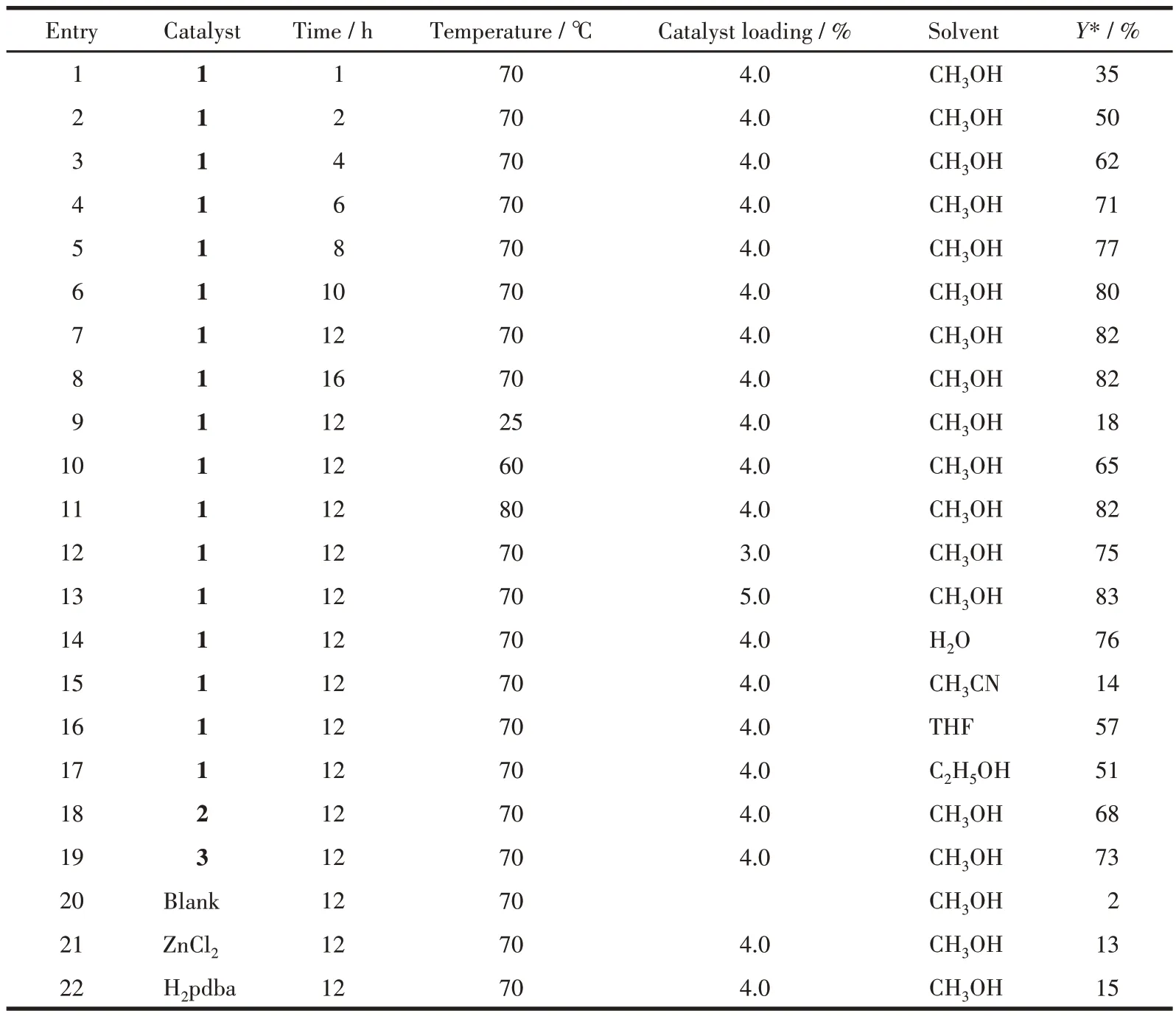
Table 1 Catalyzed Henry reaction of 4-nitrobenzaldehyde with nitromethane
Compound 1 revealed the highest activity, resulting in an 82% conversion of 4-nitrobenzaldehyde to 2-nitro-l-(4-nitrophenyl)ethan-1-ol (Table 1 and Fig.S1). Compound 1 was used to research the influence of different reaction parameters.The yield was accumulated with a yield increase from 35% to 82% on prolonging the reaction from 1 to 12 h (Table 1, Entry 1-7).The influence of catalyst amount was also investigated,revealing a product yield growth from 75% to 83% on increasing the loading of the catalyst from 3% to 5% of molar fraction (Entry 11-13). Entry 10 showed that 4% of compound 1 was able to catalyze the reaction and the yield of the product was 65% at 60 ℃. However, a mild elevated temperature of 70 ℃led to a yield of 82%. In addition to methanol, other solvents were tested. Water, ethanol, acetonitrile, and chloroform were less suitable(14%-76% yields,respectively).
In comparison with 1, compounds 2 and 3 were less active, resulting in the maximum product yields in a range of 68%-73% (Entry 18 and 19, Table 1). It should be highlighted that under similar reaction conditions, the Henry reaction of 4-nitrobenzaldehyde was significantly less efficient in the absence of the catalyst(only 2% yield) or when using H2pdba (15% yield) or ZnCl2(13% yield) as catalysts (Entry 20-22, Table 1).Although there is no clear connection between the activity and structure of the catalyst, the superior performance of compound 1 might be related to the existence of open Zn sites[16,25-26].
Different substituted benzaldehyde substrates were used to study the substrate scope in the Henry reaction with nitromethane. These tests were run under optimized conditions (x1=4.0%, CH3OH, 70 ℃, 12 h).The corresponding products were obtained in the yields varying from 15% to 82% (Table 2). Benzaldehydes containing a strong electron-withdrawing group (e.g.,nitro, and chloro substituent in the ring) revealed the best efficiency (Entry 2~5, Table 2), which can be explained by an increased electrophilicity of substrates. The benzaldehydes possessing an electron -donating functionality (e.g., methyl or methoxy group)led to lower product yields(Entry 7 and 8,Table 2).
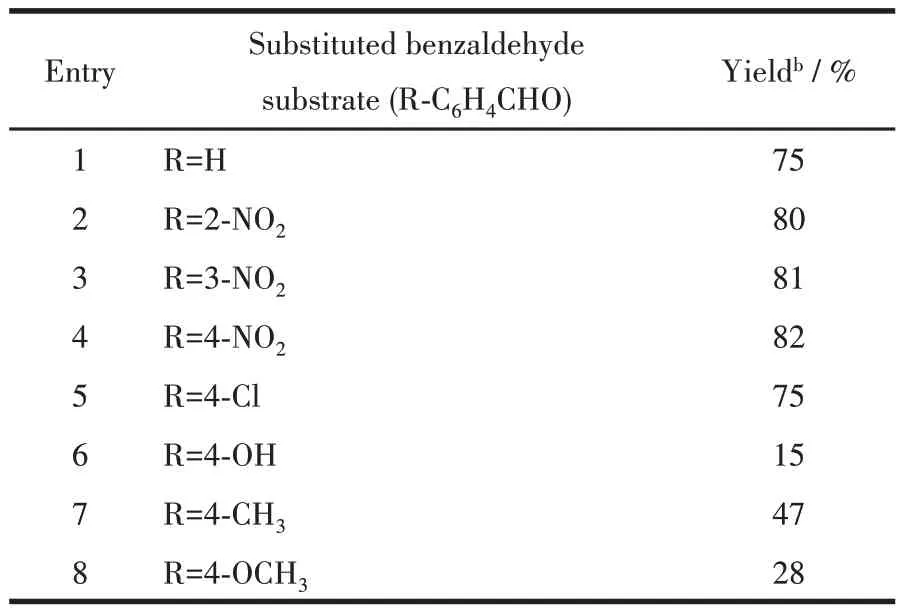
Table 2 Henry reaction of various aldehydes with nitromethane catalyzed by compound 1a
Finally, the recyclability of catalyst 1 was tested.After each reaction cycle, the catalyst was separated via centrifugation, washed with CH3OH, dried in air at about 25 ℃,and reused in the next cycle.The obtained results proved that compound 1 preserved the activity for at least five reaction cycles (the yields were 80%,79%,78%,and 76% for the second to fifth run,respectively). Besides, the PXRD patterns and the IR spectra confirm that the structure of 1 was maintained (Fig.S2 and S3), despite the appearance of several additional signals or the widening of some peaks. These alterations might be expected after a few catalytic cycles and are explained by the presence of some impurities or the decrease in crystallinity. In a model reaction involving 4-nitrobenzaldehyde and nitromethane as substrates,the catalytic performance of 1 was comparable to other heterogeneous catalytic systems based on metalcarboxylate coordination compounds (Table S4) or superior if reaction time is taken into consideration[27-31].
3 Conclusions
In summary, we have synthesized three Zn(Ⅱ), Ni(Ⅱ), and Co(Ⅱ)coordination polymers based on a dicarboxylate ligand. Compounds 1-3 disclose a 1D double chain and 2D network,respectively.The catalytic properties of these compounds were investigated. Compound 1 revealed an effective catalytic activity in the Henry reaction at 70 ℃.
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
西南医科大学学报(2022年6期)2022-11-27
高等理科教育(2022年3期)2022-06-29
高等理科教育(2022年2期)2022-05-09
化工职业技术教育(2021年5期)2021-11-09
化工职业技术教育(2021年1期)2021-03-15
高等理科教育(2020年2期)2020-06-09
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
化工职业技术教育(2020年4期)2020-01-19
衡阳师范学院学报(2016年3期)2016-07-10
化工学报(2016年3期)2016-03-14
