代谢改造大肠杆菌合成8-异戊烯基山奈酚
2023-12-18 08:13赵婉莹周景文侯颖
食品与发酵工业 2023年23期
赵婉莹,周景文,侯颖*
1(天津科技大学 生物工程学院,天津,300000)2(江南大学 生物工程学院,江苏 无锡,214122)
异戊烯基化类黄酮属于类黄酮类物质[1],其以类黄酮为骨架通过C—C键或C—O键与亲脂性的异戊烯基侧链相连构成[2],而异戊烯基修饰对提高类黄酮物质的生物活性具有重要意义[3]。8-异戊烯基山奈酚是山奈酚8-C异戊烯基化的衍生物,主要存在于豆科植物(苦参)和小檗科植物(淫羊藿)[4-5]。8-pk具有多种生物活性,如抗氧化、保护心血管[6]和降血糖[7]等。目前8-pk主要通过植物提取、化学合成和生物合成得到。相比于天然植物提取和化学合成,生物合成异戊烯基类黄酮具有反应条件温和、操作简单、选择性高和环境友好等优点。因此开发绿色高效的生物合成8-pk体系符合当今中国的“双碳”政策和可持续发展理念。
异戊烯基转移酶是黄酮类化合物异戊烯基化修饰过程中的关键酶,其自身具有的膜定位信号肽可将异戊烯基转移酶定位于细胞中不同的细胞器或细胞膜等亚细胞结构[8]。本研究团队在前期挖掘了淫羊藿来源的EkF8DT,能够识别山奈酚(kaempferol, Kae)进而合成8-pk,这为8-pk的高效生物合成提供了基础。现有的研究已在酿酒酵母中首次实现了8-pk的从头合成,通过截短N端信号肽和强化甲羟戊酸途径(mevalonate pathway,MVA)等方式,可生产25.9 mg/L的8-pk[9]。但其存在发酵周期长、生产效率低、生产成本高等问题,难以实现工业化生产。E.coli作为实验室和工业常用的菌株,因其生长迅速、技术操作简单和遗传工具发展成熟的优势而成为良好的底盘菌株。
异戊烯基转移酶可以从不同的供体来获得异戊烯基,其中以二甲基丙烯基二磷酸(dimethylallyl pyrophosphate,DMAPP)为主要的供体。DMAPP可由MVA途径和甲基赤藓糖醇4-磷酸途径(methylerythritol 4-phosphate pathway,MEP)合成。E.coli中DMAPP通过MEP途径产生,但是该途径代谢强度较弱,导致DMAPP供给不足,限制了产物的高效合成。异戊烯醇利用途径(isopentenol utilization pathway,IUP)可以分别以3-甲基-3丁烯-1-醇或3-甲基-2丁烯-1-醇作为原料,分2步生成异戊烯基二磷酸(isopentenyl diphosphate,IPP)和DMAPP(图1)。IUP途径由酿酒酵母来源的胆碱激酶(choline kinase fromSaccharomycescerevisiae,ScCK)、拟南芥来源的异戊烯磷酸激酶(isopentenyl phosphate kinase fromArabidopsisthaliana,AtIPK)和异戊烯焦磷酸异构酶(isopentenyl-pyrophosphate delta isomerase,IDI)组成,ATP是其唯一辅因子。研究表明通过表达胆碱激酶(choline kinase,CK)和磷酸异戊烯基激酶(isopentenyl phosphate kinase,IPK)引入了IUP途径使β-胡萝卜素产量增加23%[10]。通过对比IUP途径与MVA途径发现,IUP能够快速获得IPP和DMAPP,从而将IPP和DMAPP水平提高15.7倍[11]。而MVA途径会消耗大量的能量,因此,通过引入IUP途径来提高DMAPP的供给,是高效生物合成8-pk的关键。此外,将异戊烯基转移酶进行N端的截短是提高其催化性能的常见手段[12]。通过截短前40和80个氨基酸残基使得8-异戊烯基柚皮素的产量提高了44.2%和119.1%[13]。
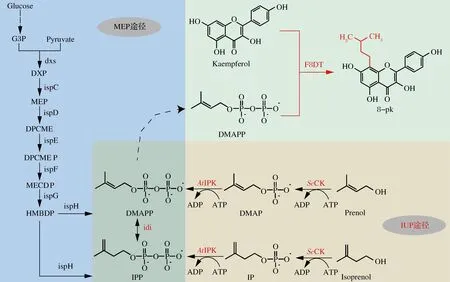
G3P:3-磷酸甘油醛;dxs:1-脱氧木酮糖-5-磷酸合成酶;ispC:1-脱氧木酮糖-5-磷酸还原异构酶;ispD:4-二磷酸胞苷-2C-甲基-D-赤藓糖醇合酶;ispE:4-二磷酸胞苷-2C-甲基-D-赤藓糖醇激酶;ispF:2C-甲基-D-红细胞-2,4-环二磷酸合酶;ispG:1-羟基-2-甲基-2-(E)-丁烯基-4-二磷酸合酶;ispH:1-羟基-2-甲基-2(E)-丁烯基-4-二磷酸还原酶;idi:焦磷酸异戊烯异构酶;DXP:1-脱氧-D-酮糖-5-磷酸盐;MEP:2-C-甲基-D-赤藓糖醇4-磷酸;DPCME:4-二磷酸胞苷-2-甲基-D-赤藓糖醇;DPCMEP:2-磷酸-4-二磷酸胞苷-2-甲基-D-赤藓糖醇;MECDP:2-C-甲基-D-赤藓糖醇-2,4-环二磷酸;HMBDP:1-羟基-2-甲基丁烯基-4-二磷酸;AtIPK:来自拟南芥的异戊烯基激酶;ScCK:来自酿酒酵母的胆碱激酶;F8DT:异戊烯基转移酶;IPP: 异戊烯基二磷酸;DMAPP:二甲基烯丙基二磷酸;IP:异戊烯基一磷酸;DMAP:一磷酸二甲基烯丙基。
针对上述问题,本文在EkF8DT游离表达的基础上,引入IUP途径,以提高DMAPP的供给;然后通过在线网站预测EkF8DT的结构,选取了8个位点进行截短,验证了其对8-pk合成的影响;最后,通过在N端融合蛋白质标签以及对质粒载体拷贝数、发酵温度、异丙基-D-硫代半乳糖苷(isopropyl-D-thiogalactoside,IPTG)浓度、诱导时机以及底物Kae的添加量优化,进一步提高8-pk的产量。
1 材料与方法
1.1 材料
1.1.1 菌株和质粒
EscherichiacoliJM109用于质粒构建与扩增。EscherichiacoliBL21(DE3)用于蛋白的表达。基因(EkF8DT,登录号QXN66318.1;Scck,登录号CP046092.1;Atipk,登录号NP_173986.2)由上海生工合成。质粒表达载体由实验室保藏。文中所使用的菌株列于表1。

表1 本实验所使用的菌株和质粒
1.1.2 培养基
LB培养基(g/L):蛋白胨10、酵母粉5、NaCl 10。
摇瓶发酵培养基(TB+葡萄糖)(g/L):甘油5、蛋白胨12、酵母提取物24、KH2PO42.31、K2HPO416.37、葡萄糖10。
5 L发酵罐:种子液按照4%(体积分数)的接种量接种于含有2.5 L TB培养基的5 L发酵罐中。初始搅拌速度300 r/min,空气流速2 vvm,温度37 ℃。发酵10 h后,将补料培养基以5 mL/h的流速加入发酵罐中,共加入500 mL。用氨水调控pH为7,设置搅拌桨转速300~600 r/min。补料培养基:500 g/L甘油。
1.2 实验方法
1.2.1 质粒和菌株的构建
所有片段均由高保真DNA聚合酶扩增,使用上海生工即用型无缝克隆试剂盒进行质粒构建。用引物对pACYC-F/pACYC-R构建质粒pACYCDuet的线性化载体,用引物对IDI-F/IDI-R,并以E.coli基因组为模板,扩增idi基因片段,通过Gibson构建质粒pACYCDuet-F8DT-idi。其他质粒的构建方法同上。本试验所用引物见表2。

表2 本实验所引用的引物
1.2.2 培养条件
采用LB培养基培养种子液。用摇瓶发酵培养基发酵生产8-pk。挑取阳性转化子于装有5 mL LB(添加相应抗性)的50 mL摇瓶中,220 r/min、37 ℃培养8~12 h。将种子液按照2%(体积分数)的接种量接种于装有25 mL摇瓶发酵培养基的250 mL摇瓶中,220 r/min,37 ℃培养,当OD600达到0.8~1时,加入终浓度为0.2 mmol/L的IPTG,温度调至25 ℃,诱导10 h后加入终质量浓度为250 mg/L的Kae,继续发酵36 h。
1.2.3 样品处理及液相分析条件
发酵结束后,取500 μL发酵液于1.5 mL离心管中并加入同体积的甲醇,剧烈振荡混匀后,12 000 r/min离心2 min,取上清液后用0.22 μm有机相滤膜过滤后使用岛津LC-20AT高效液相色谱仪进行产物检测。采用Thermo Fisher C18色谱柱(4.6 mm×250 mm,5 μm)进行色谱分离;柱温箱温度40 ℃;进样量10 μL;流速1.0 mL/min。流动相分别为:A相为超纯水(加入0.1%三氟乙酸),B相为乙腈(加入0.1%三氟乙酸);使用以下梯度实现分离:0~10 min(10%~40%B)、10~35 min(40%~80%B)、35~37 min(80%~10%B)、37~40 min(10%B)。在350 nm波长处检测产物8-pk。
2 结果与分析
2.1 异戊烯醇利用途径(IUP途径)增加DMAPP的供给
将pACYCDuet-EkF8DT转化至E.coliBL21(DE3),37 ℃培养至OD600为0.6~0.8时加入终浓度为0.2 mmol/L的IPTG,30 ℃、220 r/min诱导表达10 h后加入250 mg/L的Kae发酵60 h。然而,HPLC检测发酵液显示无8-pk的产生。推测可能的原因是异戊烯基前体供应不足,进行前体合成途径强化的研究。如图1所示,引入IUP途径,表达酿酒酵母来源的Scck和拟南芥来源的Atipk,并且共表达E.coli内源的idi以促进IPP生成DMAPP。最终,在E.coliBL21(DE3)中表达构建的pACYCDuet-EkF8DT-idi-Scck-Atipk质粒,并在发酵过程中终浓度为25 mmol/L的3-甲基-3-丁烯-1-醇。以携带pACYC空质粒的E.coliBL21作为对照1,以仅引入Scck和Atipk,不过表达E.coli内源的idi,添加3-甲基-2-丁烯-1-醇作为对照2,以仅过表达E.coli内源的idi来强化MEP途径作为对照3。如图2所示,通过上述途径构建策略,发酵36 h时,重组菌中8-pk产量最高可达到2.14 mg/L,而在对照1、对照2和对照3中均未检测到8-pk。

图2 引入IUP途径菌株EI02生物合成8-pk的过程
上述结果表明,E.coliBL21本身无8-pk合成能力。实验组能够检测到8-pk的原因可能是3-甲基-3-丁烯-1-醇作为底物时酶的kcat/Km要大于3-甲基-2-丁烯-1-醇[14],更好地增加了异戊烯基转移酶的前体DMAPP在体内的合成量,进而产生更多的8-pk。而仅过表达E.coli内源的idi来强化MEP途径不能够为催化反应提供较多的DMAPP,因此推测DMAPP供应不足是8-pk合成的关键限制因素。
2.2 异戊烯基转移酶的截短表达
根据http://www.cbs.dtu.dk/services/TMHMM/网站的预测结果显示,EkF8DT是具有至少7个跨膜结构域的膜内源性蛋白(图3-a)。膜蛋白的大量表达可能会对细胞膜产生损害,影响蛋白的可溶性表达[15],降低其催化活性甚至失活。而截短N端信号肽可以提高酶的催化活性[12]。将EkF8DT的氨基酸序列输入TargetP 2.0在线工具,对EkF8DT的信号肽进行预测(图3-b),峰的高度代表了信号肽的可能性。结果显示EkF8DT的转运肽在第49位氨基酸。并利用在线网站(https://drug.ai.tencent.com/)对蛋白质二级结构进行预测。结果显示EkF8DT由10个α-螺旋的主体骨架和一个长侧链组成。长侧链的存在可能会对酶的结构造成影响。因此构建了8个N端氨基酸被截短的菌株,分别为截短40(EI04)、49(EI05)、56(EI06)、60(EI07)、64(EI08)、82(EI09)、96(EI10)和100(EI11)个氨基酸(图4-a)。如图4-b所示,菌株EI06、EI07和EI08可分别提高2、3和2.9倍的8-pk产量。菌株EI07的产量提高最明显(6.46 mg/L)。可能是前60个氨基酸为定位信号肽,当其被截掉时,该酶无法定位到细胞膜上,同时酶的催化能力提高。菌株EI04、EI05、EI09和EI11在合成8-pk的能力上明显下降。

a-跨膜结构域;b-信号肽
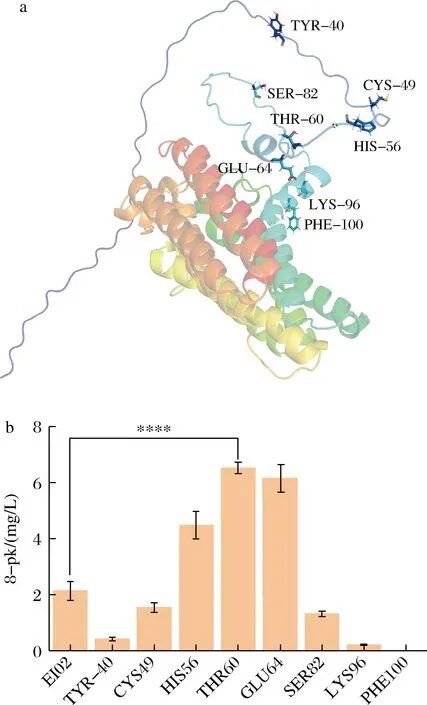
a-EkF8DT二级结构预测及截短位点;b-N端不同位点截短菌株发酵产量图
2.3 蛋白质标签改造异戊烯基转移酶
添加蛋白溶解度标签可提高外源蛋白的可溶性表达[16]。SUMO蛋白标签融合到N端时,SAM表达菌株的可溶性蛋白表达量显著提高[17]。LAI等[18]发现融合GST的蛋白在E.coliBL21(DE3)中的表达水平显著提高。NusA标签与乙醛脱氢酶融合,使得酶活性得到了提高[19]。因此我们选择了GST、NusA和SUMO增强EkF8DT的可溶性表达。如图5所示,菌株EI12的8-pk的产量较菌株EI07提高了2倍(12.92 mg/L)。而菌株EI17的产量是EI07的7.5%。结果证明SUMO标签增强了重组蛋白的表达和溶解度[20],而NusA和GST标签起到了负作用,这与之前的研究结果一致[21]。N端融合蛋白标签能够提高EkF8DT的催化能力,提高8-pk的产量,同时也证明蛋白质标签的使用具有较强的选择性。
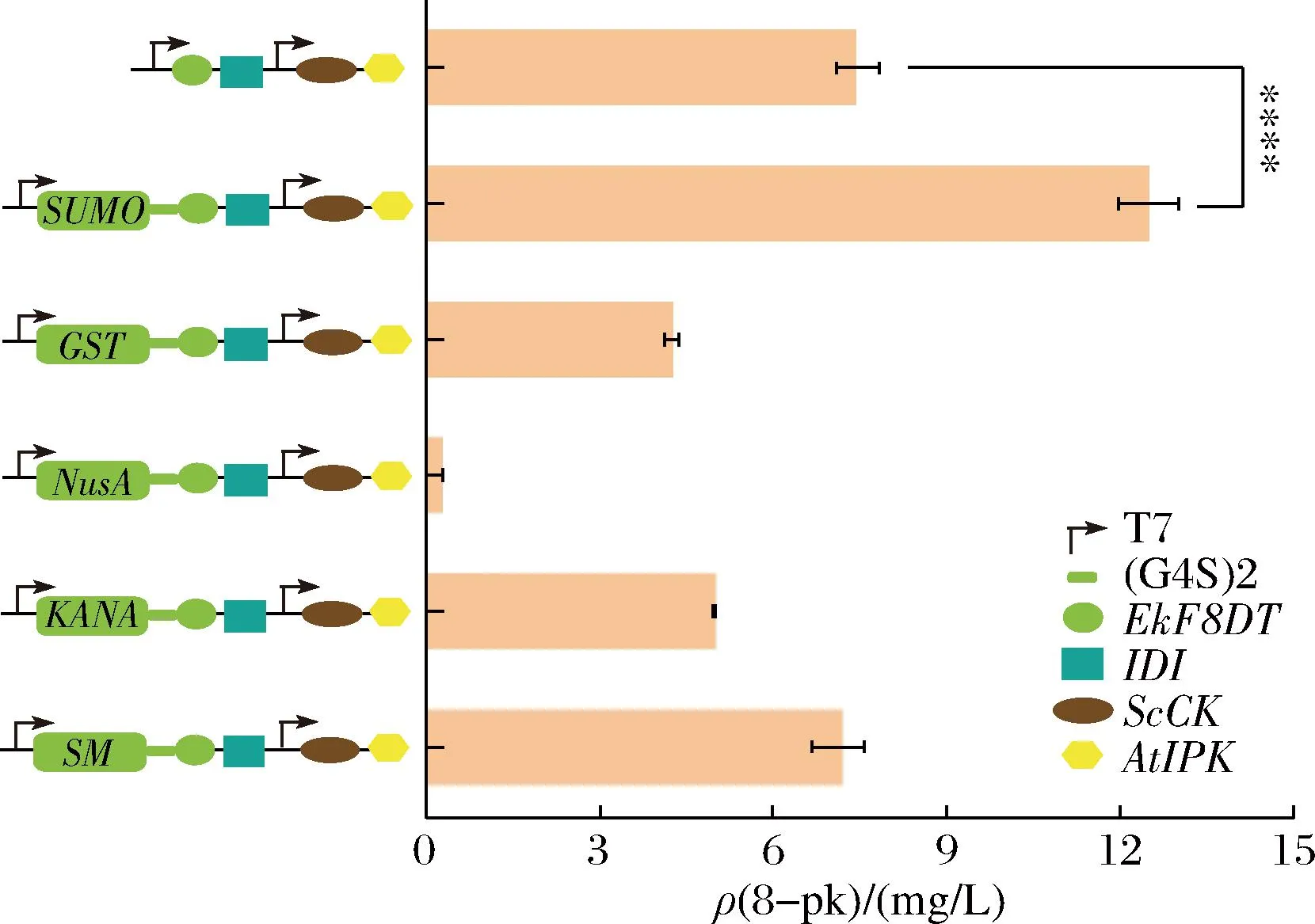
图5 N端连接不同的蛋白质标签对8-pk生物合成的影响
使用抗性基因的前导序列作为助溶标签能够增强膜蛋白的表达,如在生产香叶醇的研究中,将Sm抗性基因的前25个前导序列和KANA的前34个前导序列融合到香叶醇合酶的N末端,前者使得香叶醇的产量提高了1.3倍,后者与对照组相比无明显差异[22]。因此,将Sm和Kana的前导序列融合在EkF8DT的N端,测试结果发现菌株EI15和菌株EI16的8-pk产量为菌株EI07的97%和65%,证明该方法,无法有效提高EkF8DT的可溶性表达,降低了8-pk的合成。
2.4 质粒拷贝数对宿主细胞生长以及8-pk合成的影响
研究表明质粒拷贝数对宿主细胞生长以及产物合成有明显影响[23]。通过更换不同拷贝数的质粒载体来验证拷贝数对8-pk产量的影响。结果如图6所示,替换为质粒载体pCDFDuet、pETDuet和pRSFDuet后,8-pk产量分别为pACYCDuet为质粒载体(EI12)的63%、62%和55%,OD600分别为原菌株(EI12)54%、200%和59%。

a-不同基因拷贝数质粒构建图;b-不同拷贝数质粒菌株发酵产量图
上述研究结果表明,质粒载体拷贝数对基因表达的影响较大,虽然中拷贝质粒载体pETDuet能够使菌株菌体浓度得到较好提升,但是产量却大幅度下降,说明产量与细胞生长不呈现正相关关系。可能是由于中高拷贝质粒使得EkF8DT被表达的速度过快,出现错误的折叠方式,导致EkF8DT可溶性降低。而低拷贝质粒载体pACYCDuet会降低基因复制速度,从而降低EkF8DT折叠时的错误率,提高EkF8DT的可溶性表达量。同时对于异戊烯基转移酶膜蛋白来说,中高拷贝质粒载体使得其表达量增高,更易锚定于细胞膜上导致其活性降低,并影响菌株的生长,无法更好地行使其催化功能。
ZHANG等[24]研究发现Scck受质粒拷贝数的影响较为显著,高拷贝的质粒pRSFDuet更适合该基因在大肠杆菌中的表达及应用,因此,将IUP途径上的3个基因idi、Scck和Atipk共同构建到质粒载体pRSFDuet上,将其和pACYCDuet-SUMOEkF8DT60共同转入E.coliBL21中,得到菌株EI20,结果显示,8-pk产量仅3.60 mg/L。
2.5 8-pk合成过程的优化
在发酵过程中,诱导温度是影响细胞生长和蛋白表达的重要因素,尤其是对膜蛋白来说,在较高温度诱导表达,微生物生长速度较快,酶的合成速度也随之较快,容易形成包涵体。反之,降低诱导温度,有利于酶的正确折叠,但是细胞生长可能受影响。对菌株EI12进行发酵温度优化,在不同的诱导温度下(16、20、25、30 ℃)发酵36 h,检验8-pk产量,结果如图7-a所示,在诱导温度为25 ℃时,菌株EI12发酵生产8-pk的产量最高,为21.5 mg/L,是16、20和30 ℃发酵条件下8-pk产量的2.82、1.55和1.6倍。说明诱导EkF8DT表达的最适温度为25 ℃。

a-诱导温度;b-IPTG浓度;c-诱导时机;d-底物浓度
T7启动子的表达系统利用IPTG诱导蛋白的表达,但较高浓度的IPTG对细胞生长有负面影响[25]。在250 mL摇瓶中培养菌株EI12至OD600=0.6~0.8时,分别加入终浓度为0.05、0.10、0.15、0.20、0.25 mmol/L的IPTG,以测试不同IPTG浓度对发酵生产的影响。结果如图7-b所示,当IPTG终浓度为0.20 mmol/L时,8-pk的积累量是当IPTG终浓度为0.05、0.10、0.15、0.25 mmol/L时的5.12、2.7、1.6、1.3倍。导致该结果的原因可能是低浓度的IPTG会导致诱导不完全,蛋白表达量不够高,影响了8-pk的合成。而过高浓度的IPTG会使得转录和翻译进行的较快,增加了蛋白折叠的错误率,催化能力降低,从而影响8-pk的合成。
将种子液按照2%(体积分数)的接种量接种于装有25 mL摇瓶发酵培养基的250 mL摇瓶中,分别在OD600为0.3、0.6、0.9、1.2和1.5时加入终浓度为0.2 mmol/L的诱导剂IPTG,来验证不同的诱导时机对8-pk合成的影响。结果如图7-c所示,在OD600为0.9时加诱导剂8-pk的产量最高,为21.54 mg/L,是OD600为0.3、0.6、1.2、1.5时添加IPTG的3.9、1.4、1.2、1.9倍。
类黄酮使得E.coli的形态产生变化,损害细胞膜[26]。为确定最佳底物浓度,在添加IPTG 10 h后分别添加50、100、150、200、250 mg/L的Kae。结果如图7-d所示,底物最适质量浓度为150 mg/L,8-pk产量进一步提高至24.28 mg/L。
2.6 5-L发酵罐发酵8-pk水平验证
使用5 L发酵罐对生产菌株进行验证,探究其合成8-pk的最大潜力。进行流加补料发酵,接种5 h后开始流加补料,补料速度为5 mL/h,共补料500 mL。在OD600=29时添加终浓度为0.2 mmol/L的IPTG,诱导8 h后添加终质量浓度为250 mg/L的Kae溶液和终浓度为25 mmol/L的3-甲基-3-丁烯-1-醇溶液。结果如图8所示,发酵108 h后,8-pk的产量达到44.33 mg/L。

图8 5 L发酵罐中EI12菌株生物合成8-pk的过程
3 讨论
8-pk是一种具有高附加值的异戊烯基黄酮类化合物,其应用价值近年来逐渐被挖掘[27]。随着国家双碳战略的提出,绿色生物合成在8-pk合成中展现出极好的应用前景。关于8-pk合成的相关文献报道较少,植物来源的异戊烯基转移酶至少存在7个跨膜结构域,使其难以进行可溶性表达,以及DMAPP供应不足,这都是限制8-pk产量提高的障碍。MEP途径合成DMAPP有许多限制因素,例如G3P和丙酮酸供应的不平衡[28]和铁硫酶IspG和IspH对氧敏感而容易失活等问题[29],且MEP途径因需要辅因子的参与会与其他途径竞争资源,这种复杂的调节使得该途径提高DMAPP供应存在很多阻碍。因此,选择引入代谢途径更为简短且高效的IUP途径,可解决在E.coli中MEP途径所生产的DMAPP供应不足的问题。针对异戊烯基转移酶是膜蛋白具有定位信号肽的问题,我们采取了N端氨基酸截短的方式来解决。通过在线网站预测了信号肽位置和EkF8DT的二级结构,并根据预测的结构选取了8个代表性的位点进行截短。发现不同位置的截短会对8-pk合成产生不同效果,相比于根据预测信号肽的位置进行截短,根据AlphaFold2所预测的酶的二级结构进行截短具有更高的可信度。因此,根据酶的二级结构选择位点截短并寻找规律,来细化截短的位置是最好的选择。最后,通过融合表达蛋白质标签、优化质粒拷贝数和优化发酵条件,8-pk在5 L发酵罐的产量达到44.33 mg/L,本研究为细菌中8-pk的高效生物合成提供了一定的参考。鉴于异戊烯基转移酶具有较高的底物特异性,后期研究中通过筛选不同的酶,并结合本研究的策略可以有效地提高异戊烯基化的类黄酮类化合物的合成。
猜你喜欢
河北医学(2021年10期)2021-10-27
中国现代药物应用(2020年6期)2020-01-10
中国临床医学影像杂志(2019年6期)2019-08-27
天然产物研究与开发(2018年10期)2018-11-06
天然产物研究与开发(2018年10期)2018-11-06
中西医结合心血管病杂志(电子版)(2018年8期)2018-01-12
医学综述(2015年15期)2015-03-04
中国医学科学院学报(2015年5期)2015-03-01
发明与创新(2015年25期)2015-02-27
天然产物研究与开发(2014年6期)2014-04-27