
人参红皮病菌Rhexocercosporidium panacis 的化学成分
2023-12-11 09:37孙迪安
浙江农林大学学报 2023年6期
刘 鹏,李 琦,杨 健,孙迪安,况 燚,丁 刚
(1.浙江农林大学 化学与材料工程学院,浙江 杭州 311300;2.中国医学科学院 北京协和医学院 药用植物研究所 中草药物质基础与资源利用教育部重点实验室,北京 100193;3.中国中医科学院 国家中药资源中心 道地药材国家重点实验室培育基地,北京 100700)
真菌是天然产物的重要来源之一,目前已从中挖掘了包括抗菌药物青霉素在内的大量药源分子。随着大规模化学筛选的开展,从普通真菌资源中发现结构新颖与活性显著的次级代谢产物的概率逐渐变低。因此,独特生境来源的真菌资源成为新活性化合物研究的热点[1]。
植物病原真菌是一种潜在的植物内生真菌,一方面需抵御植物自身防御机制,另一方面需应对外界复杂环境及其他竞争者,是一类特殊生境的真菌类群,因而具有产生结构新颖化合物的潜力[2-4]。LIU 等[5]从小麦Triticumaestivum病原真菌平脐蠕孢属真菌Bipolarissp.TJ403-B1 中分离得到一系列新骨架的蛇孢假壳素(ophiobolin)类似物(bipolarolides A~G);KUMARIHAMY 等[6]从植物病原菌Septoriapistaciarum中分离得到具有抗疟、抑菌活性的化合物;LI 等[7]和WANG 等[8]从小麦病原真菌小麦根腐离蠕孢Bipolarissorokiniana中分离得到具有显著植物促生长活性和植物毒活性的倍半萜类化合物,进一步印证了植物病原真菌的代谢潜力。
人参Panaxginseng红皮病菌Rhexocercosporidiumpanacis是导致人参红皮病的病原真菌,病害主要发生在人参根部,会形成大小不一、形状不规则的红色病斑,影响人参的产量及品质,造成严重的经济损失[9-10]。目前尚未有关于人参红皮病菌次级代谢产物研究的相关报道。为探明该菌株代谢潜力,本研究选择人参红皮病菌作为研究材料,对其大米发酵产物乙酸乙酯提取部位进行化学成分研究,首次从中分离得到了9 种单体化合物,并对其抗氧化活性与植物毒活性进行初步研究。
1 材料与方法
1.1 材料
1.1.1 菌株 人参红皮病菌菌株购买自中国普通微生物菌种保藏管理中心(菌种保藏编号CGMCC3.17260)。
1.1.2 仪器与试剂 核磁共振仪:Bruker AM 600 (布鲁克科技有限公司,德国),制备型色谱柱:YMC ODS-A (250 mm×10 mm,5 μm,岛津实验器材有限公司,日本),高效液相色谱仪Agilent 1200 (安捷伦科技有限公司,美国),半制备液相色谱仪:LC-52 (赛谱锐思科技有限公司,中国),TG16-WS 高速离心机 (湘潭湘仪仪器有限公司,中国),KQ-500E 型超声波清洗器(昆山市超声仪器有限公司,中国),鼓风式干燥箱(上海一恒科学仪器有限公司,中国),暗箱式紫外分析仪(上海市宝山顾村电光仪器厂,中国)。色谱甲醇(赛默飞世尔科技,美国),石油醚、乙酸乙酯、二氯甲烷、甲醇、乙醇、丙酮等其他试剂(北京化学试剂公司,中国),制备薄层层析硅胶(上海彭腾精细化工有限公司,中国),Sephadex LH-20 (法玛西亚公司,瑞典)。
1.1.3 培养基 PDA 培养基配方:马铃薯200 g,葡萄糖20 g,琼脂粉20 g,去离子水1 000 mL;大米固体培养基配方:每60 g 大米加入80 mL 去离子水置于500 mL 锥形瓶中。
1.2 方法
1.2.1 菌株发酵 人参红皮病菌菌种接种在PDA 培养基上,20 ℃条件下静置培养10 d,再用接种针将其转接种于含大米培养基的500 mL 锥形瓶中(121 ℃,30 min 高压灭菌锅灭菌),25 ℃静置培养2 周。共发酵80 瓶。
1.2.2 化合物的提取分离 菌株发酵结束后,向锥形瓶中加300 mL 乙酸乙酯进行萃取,合并乙酸乙酯相,此操作重复3 次,减压浓缩得到乙酸乙酯部位总膏(30 g)。乙酸乙酯粗提物(30 g)经正相硅胶柱粗分划段,以石油醚∶丙酮(体积比为1∶0~0∶1)梯度洗脱,依据薄层色谱法检测合并得到8 个组分(Fr.1~Fr.8)。Fr.2(500 mg)经重结晶得到化合物1 (5 mg)。Fr.3 (2.3 g)经正相硅胶柱层析,以石油醚∶丙酮(体积比为80∶1~3∶1)继续分离得到4 个组分(Fr.3.1~Fr.3.4)。Fr.3.2 经Sephadex LH-20 (甲醇为洗脱剂)分离得5 个组分Fr.3.2.1~Fr.3.2.5,其中Fr.3.2.5 经半制备液相色谱(甲醇/水,体积分数为51%甲醇,流速2 mL·min-1)纯化得到化合物4 [3.5 mg,保留时间(tR)为20 min]。Fr.3.3 以丙酮为洗脱剂经凝胶柱 (Sephadex LH-20)分离得到7 个组分Fr.3.3.1~Fr.3.3.7,其中Fr.3.3.3 有结晶析出,通过重结晶及半制备液相(甲醇/水,体积分数为98%甲醇,流速2 mL·min-1)纯化得到化合物2 (2.4 mg,tR=19 min)和化合物3 (9.3 mg,tR=23 min)。Fr.3.3.7 经半制备液相(甲醇/水,0~5 min,体积分数为40%甲醇;5~35 min,体积分数为40%~80%甲醇,流速2 mL·min-1)纯化得到化合物6 (2.5 mg,tR=31 min)。Fr.5 (1.3 g)经正相硅胶柱层析,以二氯甲烷∶甲醇(体积比为50∶1~0∶1)梯度洗脱得Fr.5.1~Fr.5.6,其中Fr.5.3 经Sephadex LH-20 (二氯甲烷∶甲醇体积比为1∶1)分离得到Fr.5.3.1~Fr.5.3.6。Fr.5.3.5 经过Sephadex LH-20 (二氯甲烷∶甲醇体积比为1∶1)分离得Fr.5.3.5.1~ Fr.5.3.5.4,其中Fr.5.3.5.3 经半制备液相(甲醇/水,0~5 min,体积分数为50%甲醇;5~35 min,体积分数为50%~64%甲醇,流速2 mL·min-1)纯化得化合物8 (2.4 mg,tR=19 min),Fr.5.3.5.4 经半制备液相(甲醇/水,0~5 min,体积分数为20%甲醇;5~30 min,体积分数20%~55%甲醇;30~50 min,体积分数为55%~70%甲醇,流速2 mL·min-1)纯化得化合物9 (2.4 mg,tR=32 min)、化合物5 (2.4 mg,tR=44 min)。Fr.6 经Sephadex LH-20 (甲醇为洗脱剂)分离得到4 个组分Fr.6.1~Fr.6.4,其中Fr.6.3 经半制备液相处理(甲醇/水,0~5 min,体积分数为40%甲醇;5~30 min,体积分数为40%~60%甲醇,流速2 mL·min-1)纯化得到化合物7 (2.0 mg,tR=23 min)。
1.2.3 抗氧化活性评价 采用1,1-二苯基-2-苦肼基(DPPH)自由基清除试验评价化合物1~9 的抗氧化活性[11]。将待测化合物及阳性对照(维生素E,VE)配置成质量浓度梯度(200、100、80、60、40、20、10、5 mg·L-1),在96 孔板中加入60 μL 不同质量浓度的样品或VE,然后加入60 μL 的0.1 mmol·L-1DPPH 甲醇溶液,振荡均匀,在黑暗中孵育2 h 后,用酶标仪测定在517 nm 处的吸光度,空白对照为60 μL DPPH 加上60 μL 甲醇。抑制率={[D(517)blank-D(517)sample]/D(517)blank}×100%。其中:D(517)blank为对照组60 μL 甲醇与60 μL DPPH 混合溶液吸光度,D(517)sample为60 μL 样品与60 μL DPPH 混合溶液吸光度。试验重复3 次,以半清除率(IC50,DPPH 自由基清除率为50%时的样品浓度)为评价指标。
1.2.4 植物毒活性评价 参考文献[12]方法测定单体化合物4~9 对离体人参根部的致病性。具体步骤如下:采集新鲜人参根,先用自来水清洗,去除泥土并摒弃有瑕疵的根,接着用体积分数为75%乙醇对根部进行消毒,再用打孔器在根部中心位置制作3 个孔洞后,将其放置在装有滤纸片的培养皿中(滤纸片已提前用无菌水浸润)。化合物用体积分数为75%乙醇配置成质量浓度为1 g·L-1的标准溶液。最后用移液枪分别吸取10 μL 待测化合物标准溶液加入到人参3 个孔洞中,封口膜密封后,在黑暗条件下,20 ℃孵育。体积分数为75%乙醇处理的人参根作为对照组,3 d 后观察结果。
2 结果与分析
2.1 化合物结构鉴定
运用多种分离技术从人参红皮病菌粗提物的乙酸乙酯部位纯化得到9 种单体化合物(图1),利用质谱及核磁共振波谱技术并结合文献数据比对,化合物结构鉴定结果如下:化合物1 为麦角甾醇(ergosterol)、化合物2 为过氧化麦角甾醇(5,8-epidioxy-5α,8α-ergosta-6,9,22E-tien-3β-ol)、化合物3 为5,8-表二氧麦角甾醇-6,9(11),22-三烯-3-醇(5,8-epidioxy-5α,8α-ergosta-6,22E-dien-3β-ol)、化合物4 为核桃酮(regiolone)、化合物5 为4,6,8-三羟基-3,4-二氢-1(2H)-萘酮(4,6,8-trihydroxy-3,4-dihydronaphthalen-1(2H)-one)、化合物6 为2,5-二甲基-7-羟基色酮(2,5-dimethyl-7-hydroxychromone)、化合物7 为2-甲基-5-羧甲基-7-羟基色酮(2-methyl-5-carboxymenthyl-7-hydroxychromone)、化合物8 为(+)-citreoisocoumarin 和化合物9 为de-O-methyldiaporthin。化学结构式如图1 所示。
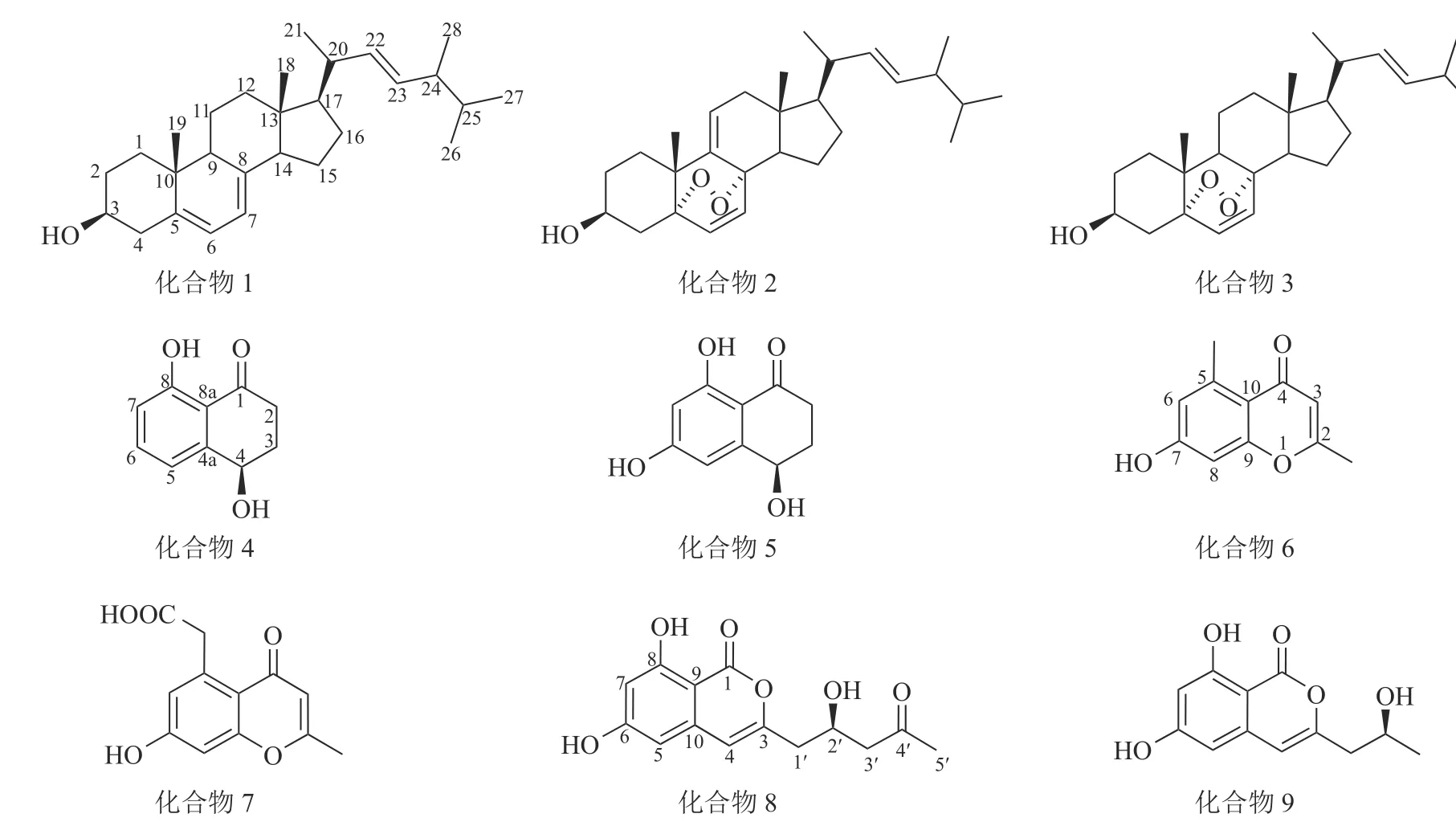
图1 化合物1~9 的化学结构Figure 1 Chemical structures of compounds 1-9
化合物1:无色晶体,基于1H-NMR 图谱分析,高场区氢化学位移(δH)在1.20~2.50 间有复杂重叠的烷基氢信号,并存在6 个甲基信号,其中2 个单峰甲基氢信号(δH=0.63 和0.95),4 个双峰甲基氢信号(δH=1.04、0.92、0.83、0.81),另在低场区化学位移δH=3.64 处有个积分为1 的多重峰氢信号,推测此处连接1 个羟基;低场区存在2 对烯烃质子信号,其中1 对δH=5.21 和5.19 间的耦合常数为15.6,推测为环外侧链反式双键,基于以上特征推测化合物1 为麦角甾醇类化合物。1H-NMR (CDCl3, 600 MHz):δH5.57 (1H, d,J=6.0 Hz, H-7), 5.38 (1H, d,J=6.0 Hz, H-6), 5.21 (1H, dd,J=15.6, 7.2 Hz, H-23), 5.19 (1H, dd,J=15.6, 7.2 Hz, H-22), 3.64 (1H, tt,J=11.4, 4.2 Hz, H-3), 1.04 (3H, d,J=6.6 Hz, H-21), 0.95 (3H, s, H-19), 0.92(3H, d,J=6.6 Hz, H-28), 0.83 (3H, d,J=6.6 Hz, H-27), 0.81 (3H, d,J=6.6 Hz, H-26), 0.63 (3H, s, H-18)。以上氢谱数据与文献[13]对比后确认此化合物为麦角甾醇。
化合物 2:无色晶体,其1H-NMR 图谱发现与麦角甾醇相似。通过与文献[14]比对,确认该化合物为过氧化麦角甾醇。1H-NMR (CDCl3, 600 MHz):δH6.58 (1H, d,J=8.4 Hz, H-6), 6.27 (1H, d,J=8.4 Hz, H-7),5.41 (1H, d,J=6.0, 1.8 Hz, H-11), 5.22 (1H, dd,J=15.6, 7.8 Hz, H-23), 5.14 (1H, dd,J=15.6, 8.4 Hz, H-22), 4.00(1H, m, H-3), 1.08 (3H, s, 19-Me), 0.99 (3H, d,J=6.6 Hz, 21-Me), 0.90 (3H, d,J=6.6 Hz, 28-Me), 0.82 (3H, d,J=6.6 Hz, 26-Me), 0.81 (3H, d,J=6.6 Hz, 27-Me), 0.72 (3H, s, 18-Me)。
化合物3:无色晶体,其1H-NMR 谱图与过氧化麦角甾醇高度相似,主要区别在化学位移δH=5.41 处的氢信号消失,推测双键被还原。通过与文献[14]比对,确认此化合物为5,8-表二氧麦角甾醇-6,9(11),22-三烯-3-醇。1H-NMR(CDCl3, 600 MHz):δH6.50 (1H, d,J=8.4 Hz, H-6), 6.24 (1H, d,J=8.4 Hz, H-7), 5.22 (1H, dd,J=15.6, 7.8 Hz, H-23), 5.14 (1H, dd,J=15.6, 8.4 Hz, H-22), 3.96 (1H, m, H-3), 0.99 (3H, d,J=6.6 Hz, 21-Me), 0.90 (3H, d,J= 6.6 Hz, 28-Me), 0.88 (3H, s, 19-Me), 0.83 (3H, d,J=6.6 Hz, 26-Me), 0.81(3H, d,J=6.6 Hz, 27-Me), 0.81 (3H, s, 18-Me)。
化合物4:黄色粉末,低分辨质谱数据质荷比(m/z)为179.07,[M+H]+。1H-NMR 谱图(图2)显示在低场区有3 个氢质子信号,δH=7.50 (1H,t,J=7.8 Hz),7.02 (1H,dd,J=7.8,1.2 Hz),6.93(1H,dd,J=7.8,1.2 Hz),推测化合物含苯环结构;低场区化学位移δH=12.89 处的尖单峰结合13C-NMR 谱图(图3)中碳化学位移(δC) = 204.4,推测为与羰基形成分子内氢键的活泼氢信号;化学位移δH=4.92 的1 个双峰氢信号暗示此处可能连接1 个羟基。另外,高场区有2 对相互耦合的亚甲基信号(δH=3.00,2.65 和δH=2.35,2.20),基于以上数据推测化合物4 为芳香类化合物。1H-NMR (CDCl3, 600 MHz):δH12.89 (1H,8-OH, s), 7.50 (1H, t,J=7.8 Hz, H-6), 7.02 (1H, dd,J=7.8, 1.2 Hz, H-5), 6.93 (1H, dd,J=7.8, 1.2 Hz, H-7), 4.92(1H, dd,J=7.8, 3.6 Hz, H-4), 3.00 (1H, ddd,J=18.0, 8.4, 4.8 Hz, H-2a), 2.65 (1H, ddd,J=18.0, 8.4, 4.8 Hz, H-2b),2.35 (1H, m, H-3a), 2.20 (1H, m, H-3b)。13C-NMR (CDCl3, 150 MHz):δC204.4 (C-1), 34.7 (C-2), 31.4 (C-3),67.9 (C-4), 146.0 (C-4a), 117.9 (C-5), 137.1 (C-6), 117.5 (C-7), 162.9 (C-8), 115.4 (C-8a)。以上核磁数据与文献[15]中的核桃酮基本一致。

图2 化合物4 的1H-NMR 谱图(600 MHz,CDCl3)Figure 2 1H-NMR spectrum (600 MHz) of 4 in CDCl3
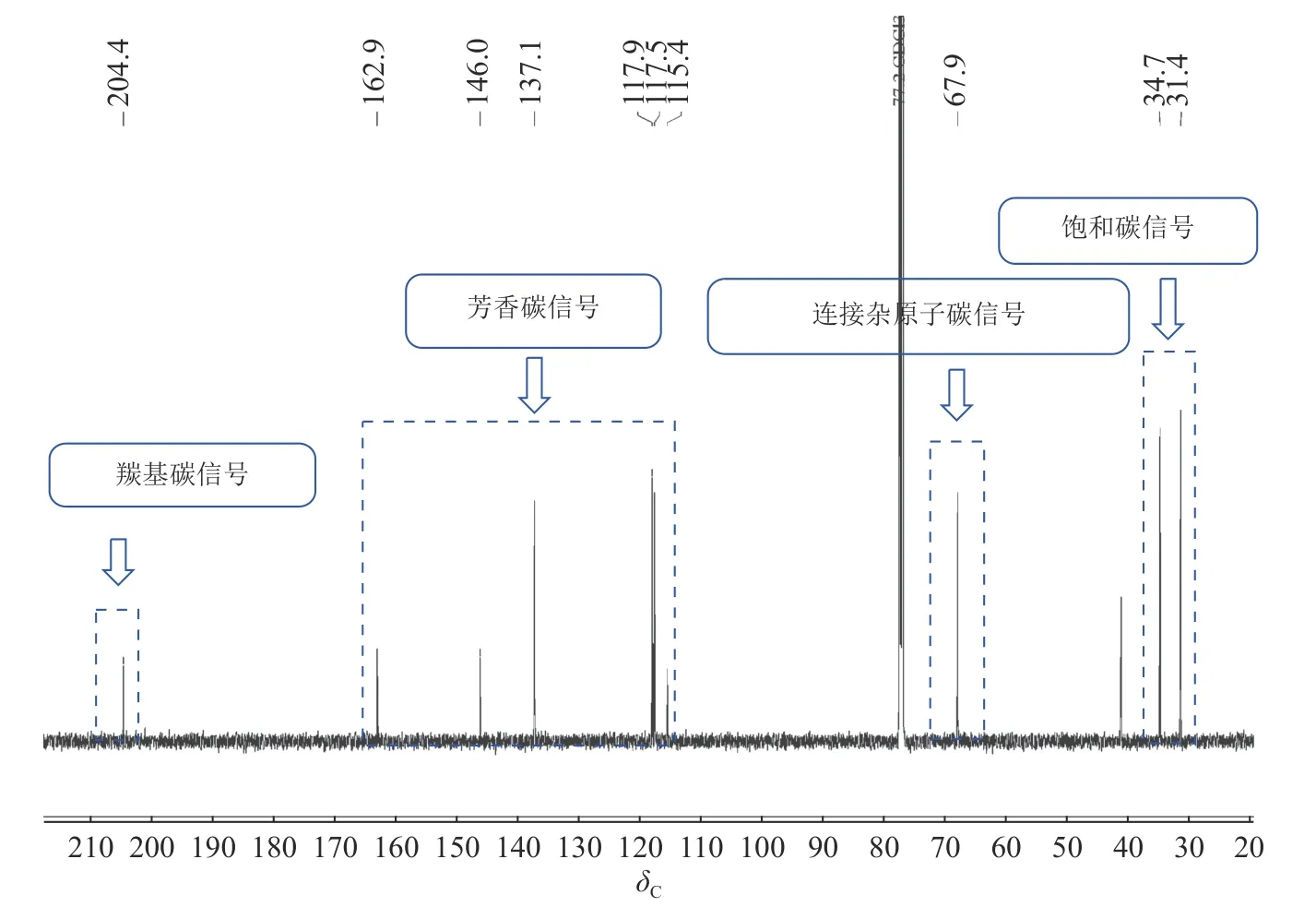
图3 化合物4 的13C-NMR 谱图(150 MHz,CDCl3)Figure 3 13C-NMR spectrum (150 MHz) of 4 in CDCl3
化合物5:黄色粉末,低分辨质谱数据m/z=195.09,[M+H]+。化合物5 的1H-NMR 谱图与化合物4 高度相似,主要区别在化学位移δH=6.00~8.00 范围内少1 个芳香氢信号,且剩下2 个芳香氢δH=6.64 和6.22 之间耦合常数为1.8,判断这2 个氢在苯环上处于间位。对比化合物4,化合物5 的芳香氢化学位移向高场移动,结合碳谱数据分析,发现化合物5 比化合物4 多了1 个δC在166.5 处的碳信号,推测为双键连氧的碳信号,即苯环上新增的取代基为羟基。1H-NMR(Acetone-d6, 600 MHz):δH12.89 (1H, s, 8-OH), 6.64 (1H, d,J=1.8 Hz, H-5), 6.22 (1H, d,J=1.8 Hz, H-7), 4.78 (1H, dd,J=9.0, 4.2 Hz, H-4) ,2.75 (1H, m,J=17.4, 6.0, 4.8 Hz, H-2a), 2.61 (1H, m, H-2b ), 2.26 (1H, m, H-3a), 2.03 (1H, m, H-3b)。13C NMR(Acetone-d6, 150 MHz):δC203.1 (C-1), 166.5 (C-6), 166.1 (C-8), 151.5 (C-4a), 109.9 (C-8a), 106.9 (C-5),102.1 (C-7), 68.0 (C-4), 35.6 (C-2), 32.7 (C-3)。以上核磁共振数据与文献[16]中的4,6,8-三羟基-3,4-二氢-1(2H)-萘酮基本一致。
化合物6:黄色粉末,低分辨质谱数据m/z=191.10,[M+H]+。1H-NMR 谱图显示在低场区δH=6.68和6.67 处有2 个互相耦合的氢信号,结合耦合常数2.4 可知为苯环上处于间位的氢,表明化合物6 存在被4 个取代基取代的苯环结构;化学位移δH=5.92 推测为单峰烯烃质子信号,结合13C-NMR 谱图中δC=179.3、164.5 和117.5 等3 处的碳化学位移推测化合物含α,β-不饱和酮结构;高场区存在2 个单峰甲基氢信号(δH=2.70 和2.28),根据化学位移值推测这2 个甲基分别连接在苯环和双键上。基于以上数据推测化合物6 为色酮类化合物。1H-NMR(Acetone-d6, 600 MHz):δH9.37 (7-OH, s), 6.68 (1H, d,J=2.4 Hz, H-8), 6.67 (1H, d,J=2.4 Hz, H-6), 5.92 (1H, s, H-3), 2.70 (3H, s, 5-CH3), 2.28 (3H, s, 2-CH3)。13C-NMR(Acetoned6, 150 MHz):δC179.3 (C-4), 164.5 (C-2), 161.5 (C-7), 158.4 (C-9), 142.4 (C-5), 117.5 (C-3), 115.0 (C-10),111.8 (C-6), 101.5 (C-8), 22.8 (5-CH3), 19.7 (2-CH3)。以上核磁共振数据与文献[17]中的2,5-二甲基-7-羟基色酮基本一致。
化合物7:黄色粉末,低分辨质谱数据m/z=235.09,[M+H]+。化合物7 的1H-NMR 谱图与化合物6 的相似,主要区别在于苯环上取代的甲基信号(δH=2.70)消失,多了1 个单峰亚甲基信号(δH=4.10),结合13C-NMR 谱图新出现的信号δC=175.9,推测此处由甲基取代变为羧甲基取代。1H-NMR (CD3OD, 600 MHz):δH6.75 (1H, d,J=2.4 Hz, H-8), 6.69 (d,J=2.4 Hz, 1H, H-6), 6.01 (1H, s, H-3), 4.10 (2H, s, 5-CH2), 2.34(3H, s, 2-CH3)。13C-NMR(CD3OD, 150 MHz):δC167.5 (C-2), 119.3 (C-3), 181.5 (C-4), 115.2 (C-4a), 139.6 (C-5), 175.9 (-COOH), 111.7 (C-6), 163.7 (C-7), 102.8 (C-8), 160.7 (C-8a), 21.1 (2-CH3), 42.3 (5-CH2)。以上核磁共振数据与文献[18]中的2-甲基-5-羧甲基-7-羟基色酮基本一致。
化合物8:无色晶体,低分辨质谱数据m/z=279.13,[M+H]+。基于1H-NMR 谱图分析,低场区(δH=6.41 和6.37)处有2 个互相耦合的氢信号,耦合常数为2.4,推测为苯环上间位取代的氢。化学位移(δH=11.14)处存在1 个积分为1 的氢信号,推测为形成分子内氢键的活泼氢信号,13C-NMR 谱图(δC=167.1)处的碳信号可能为酯羰基信号,结合烯烃单峰质子信号(δH=6.41)推测化合物8 为异香豆素类结构。1H-NMR(Acetone-d6, 600 MHz):δH11.14 (1H, s, 8-OH), 6.43 (1H, s, H-4), 6.41 (1H, d,J=2.4 Hz, H-5),6.37 (1H, d,J=2.4 Hz, H-7), 4.47 (1H, tt,J=7.8, 4.8 Hz, H-2′), 2.70 (1 H, m, H-3′a, overlapped), 2.70 (1H, m,H-1′a, overlapped), 2.68 (1H, m, H-3′b), 2.60 (1H, dd,J=14.4, 8.4 Hz, H-1′b), 2.15 (3H, s, 5′-CH3),13CNMR:(Acetone-d6,150 MHz):δC167.1 (C-1), 155.7 (C-3), 106.8 (C-4), 103.6 (C-5), 167.1 (C-6, overlapped),102.5 (C-7), 164.5 (C-8), 99.6 (C-9), 140.8 (C-10), 41.9 (C-1′), 66.1 (C-2′), 50.7 (C-3′), 207.5 (C-4′), 30.6 (C-5′)。以上核磁共振数据与文献[19]中的(+)-citreoisocoumarin 基本一致。
化合物9:无色晶体,低分辨质谱数据m/z=237.11,[M+H]+。化合物9 的1H-NMR 谱图与化合物8 高度相似,表明为同类型化合物,主要区别在化合物9 的1H-NMR 谱图中少了1 个单峰甲基信号(δH=2.15),多了1 个双峰甲基信号,结合碳谱数据对比发现化合物9 比化合物8 少了2 个碳信号,其中包括1 个羰基碳信号(δC=207.5),推测化合物9 比化合物8 少了侧链上的乙酰基。1H-NMR(Acetone-d6,600 MHz):δH11.16 (1H, s, 8-OH), 6.43(1H, s, H-4), 6.41(d,J=1.8 Hz, 1H, H-5), 6.37(1H, d,J=1.8 Hz, H-7),4.17(1H, m, H-2′), 2.61(1H, dd,J=14.4, 4.8 Hz, H-1′a), 2.56(1H, dd,J=14.4, 7.8 Hz, H-1′ b), 1.23(3H, d,J=6.0 Hz, 3′-CH3)。13C-NMR(Acetone-d6, 150 MHz):δC167.1 (C-1), 156.5 (C-3), 106.4 (C-4), 103.4 (C-5),166.5 (C-6), 102.3 (C-7), 164.5 (C-8), 99.8 (C-9), 140.9 (C-10), 43.9 (C-1′), 65.5 (C-2′), 23.7 (C-3′)。以上核磁共振数据与文献[20]中的de-O-methyldiaporthin基本一致。
2.2 活性结果
抗氧化实验结果表明:与对照组VE 相比,化合物1~9 的DPPH 自由基清除效果不显著,无抗氧化活性(表1)。人参根部植物毒活性实验结果发现:化合物4~9 处理的人参根部与对照组(体积分数为75%乙醇)对比无明显差异(图4),表面均无病斑出现,表明化合物4~9 对人参根部无植物毒活性。
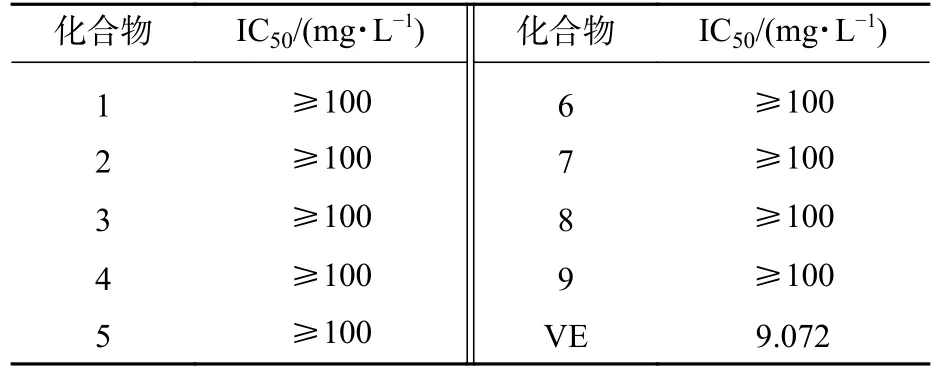
表1 化合物1~9 的抗氧化活性Table 1 Antioxidant activity of compounds 1-9

图4 化合物4~9 对人参根部致病性实验结果Figure 4 Results of pathogenicity of compounds 4-9 on roots of ginseng
3 讨论
本研究对人参红皮病菌次级代谢产物进行了初步研究。通过多种色谱分离技术首次从人参红皮病菌的大米发酵物中分离得到9 种化合物,并通过质谱分析、核磁共振波谱分析等技术对化合物结构进行了鉴定。其中化合物1~3 为麦角甾醇类化合物,4~9 为聚酮类化合物。文献报道[21]:5,8-表二氧麦角甾醇-6,9(11),22-三烯-3-醇对脂多糖(LPS)诱导巨噬细胞RAW264.7 产生一氧化氮(NO)具有中等抑制作用,表明该化合物具有一定抗炎活性。核桃酮和4,6,8-三羟基-3,4-二氢-1(2H)-萘酮都属于萘酮类化合物,其中核桃酮对莴苣Latucasativa、萝卜Raphanussativus、黄瓜Cucumissativus、洋葱Alliumcepa和小麦种子萌发和幼苗生长的植物毒性研究结果表明:在低浓度时会刺激生长,高浓度时具有抑制作用[22];而4,6,8-三羟基-3,4-二氢-1(2H)-萘酮从多种真菌的次级代谢产物中分离得到过[23-26],该化合物具有一定的抗真菌和抗细菌活性,对白色念珠菌Candidaalbicans和枯草芽孢杆菌Bacillussubtilis有中等抑制作用[27]。2009 年周永平等[16]首次报道了萘酮类化合物核桃酮和4,6,8-三羟基-3,4-二氢-1(2H)-萘酮具有杀线虫活性。2,5-二甲基-7-羟基色酮与2-甲基-5-羧甲基-7-羟基色酮属于色酮类化合物,结构中的色酮环常作为关键药效基团应用于药物设计[27]。2,5-二甲基-7-羟基色酮还具有抑制生物膜形成的能力,对金黄色葡萄球菌Staphylococcusaureus和枯草芽孢杆菌等革兰氏阳性菌有强抑制作用(抑制率达70%~80%),对大肠埃希菌Escherichiacoli和铜绿假单孢菌Pseudomonasaeruginosa等革兰氏阴性菌有中等抑制作用(抑制率为40%~60%)[28]。(+)-citreoisocoumarin 具有抗菌活性,能抑制金黄色葡萄球菌、白色念珠菌和大肠埃希菌的生长;四甲基偶氮唑盐比色法(MTT 法)评价结果表明:(+)-citreoisocoumarin 对2 株癌细胞系(MDA-MB-231,HCT 116)具有中等抑制作用[29]。本研究也评价了所分离化合物的抗氧化活性和植物毒活性,结果显示:化合物1~9 未表现出抗氧化活性,所测化合物4~9 对离体人参根部没有植物毒活性。
4 结论
本研究虽然未从人参红皮病菌发酵物中获得对人参根部有植物毒活性的化合物,但从中分离得到的化合物具有多种生物活性,表明该菌株仍具有发现活性化合物的潜力。未来,可以通过大规模发酵或改变发酵条件,进行深入的化学成分分析、分离与结构鉴定,以挖掘得到更多活性化合物,丰富该菌的次级代谢产物种类,也为人参红皮病菌病害致病因子研究提供研究基础。
猜你喜欢
基层中医药(2021年7期)2021-11-02
家庭百事通(2020年7期)2020-08-02
海峡姐妹(2019年8期)2019-09-03
中成药(2018年2期)2018-05-09
红蜻蜓·低年级(2017年6期)2017-10-30
广东第二课堂·小学(2016年11期)2016-12-06
新乡学院学报(2016年6期)2016-12-01
华人时刊(2016年13期)2016-04-05
当代化工研究(2016年9期)2016-03-20
中国蔬菜(2015年9期)2015-12-21
