
黏多糖贮积症Ⅲ型基因突变谱及造血干细胞移植效果分析
2023-12-10 13:23:06乔广明
现代中西医结合杂志 2023年19期
刘 军,刘 芳,乔广明
(1. 解放军联勤保障部队第九八〇医院,河北 石家庄 050082;2. 河北中西医结合儿童医院,河北 石家庄 050000)
黏多糖贮积症(mucopolysaccharidoses,MPS)是一类罕见的由遗传缺陷引起的溶酶体贮积病(lyso-somal storage diseases,LSD),也是造成儿童发育迟缓、认知能力下降等神经系统异常遗传性疾病之一[1]。这些遗传缺陷导致了参与糖胺聚糖(glycosaminoglycans,GAGs) 降解的酶缺乏或活性不足,GAGs是长而无分支的多糖,在细胞黏附和细胞信号传导等过程中起作用。未降解的GAGs被认为是MPS的主要和直接原因,而GAG的贮积可导致细胞的二次和三级效应,如自噬、凋亡和线粒体功能障碍[2],GAGs可积聚在细胞溶酶体中,导致受累组织功能障碍,引起面部粗糙、认知迟缓、肝脾肿大、疝、脊柱后凸、角膜混浊等多器官严重症状[3-4]。MPS分为7个亚型,其中MPSⅢ型又称Sanfilippo综合征,属于常染色体隐性遗传病,发病率约为1/1 000 000~9/1 000 000活产。在线《人类孟德尔遗传》数据库中,MPSⅢ型包含A型、B型、C型、D型4个亚型,其特征分别为缺乏硫酸乙酰肝素硫酸酯酶(heparan-N-sulfatase,SGHS)、N-乙酰-α氨基葡萄糖苷酶(α-N-acetyglucosaminidase,NAGLU)、乙酰CoA-氨基葡糖-N-乙酰转移酶(α-glucosaminidase acetyltransferase,HGSNAT)和N-乙酰氨基葡萄糖-6-硫酸酯酶(N-acetylglucosamin-6-sulfatase,GNS),这些酶都参与硫酸乙酰肝素(heparan sulfat,HS)的降解,酶的缺乏会导致HS在溶酶体内的贮积[1-5]。MPS是一种进展性疾病,诊断需要结合临床表现、影像学检查,酶学测定是MPS诊断的金标准[1]。治疗MPS的主要方法是酶替代治疗(enzyme replacement therapy,ERT)和造血干细胞移植(hematopoietic stem cell transplantation,HSCT)[6-7]。由于ERT治疗价格昂贵,临床上HSCT仍然是医生选择的一种手段[8]。目前对HSCT治疗MPSⅡ型效果肯定[7],但是对HSCT治疗MPSⅢ型仍存在争议[5],国内鲜见HSCT治疗MPSⅢ型报道。本文回顾性分析了5例MPSⅢ型患儿的临床特征、基因变异类型及HSCT治疗效果及预后,旨在加深临床医师对MPSⅢ型的认识。
1 资料与方法
1.1一般资料 选取2021年1月—2022年12月在河北中西医结合儿童医院血液科就诊的5例可疑MPS患儿,其就诊目的均希望接受HSCT治疗。在就诊于河北中西医结合儿童医院血液科前,1例曾就诊于华西医科大学二院,1例曾就诊于西安儿童医院,1例曾就诊于首都儿研所。5例患儿依据临床症状、影像学检查、血生化检查、酶活性测定及基因分析进行综合诊断,MPSⅢ型诊断参照Stapleton等[9]报道标准。5例患儿确诊后接受了HSCT治疗。本研究经河北中西医结合儿童医院伦理委员会和解放军联勤保障部队第九八○医院伦理委员会批准(2023-KY-32)。所有患儿的父母均签署了临床检查及治疗书面知情同意书。其中基因分析的判读解释由解放军联勤保障部队第九八○医院完成,HSCT治疗由河北中西医结合儿童医院完成。
1.2全外显子测序 在家长知情同意的前提下,采用EDTA抗凝管抽取5例患儿及其父母静脉血2 mL,1例送康旭检验公司,1例送贝瑞基因检测公司,3例送迈基诺基因检测公司。上述公司均采用的是全外显子捕获技术,建库后进行高通量测序。使用BWA软件(0.5.9,http://bio-bwa.sourceforge.net/)比对测序序列与GRCh37/hg19人类参考基因组。使用GATK软件(版本 4.1.7, https://software.broadinstitute.org/gatk/)分析单核苷酸变异、小片段插入及缺失等。 根据2015年美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)发布的变异解读标准和指南进行致病性评估[10]。
1.3白细胞SGHS及HGSNAT活性检测 经患儿家长知情同意,5例患儿在河北中西医结合儿童医院采静脉血4 mL,收集到含有EDTA抗凝剂的真空采血管中,将标本送至北京中科医学检验实验室。血样标本加入预处理试剂,通过低渗裂解全血中的红细胞,离心分离获得白细胞。用考马斯亮蓝法对标本白细胞蛋白浓度进行标定,用荧光底物法进行SGHS活性及HGSNAT酶活性的测定。样本加入反应体系中37 ℃恒温水浴中温育48 h,使用F93荧光分光光度计于Ex=365 nm/Em=444 nm条件下测定特异荧光值。配制4-MU标准液,制作标准曲线,随标本同时测定荧光值。计算酶活性,以nmol/(g·h)为单位来表示酶活性。
1.4HSCT治疗 5例患儿均采用了异基因非血缘脐带血干细胞移植,脐带血干细胞来源于国家卫健委批准的国家脐带血库,HLA配型为8/10~9/10相合,预处理方案BU+CY+ALG+FLU抗人T细胞猪免疫球蛋白(ALG 武汉中生毓普生物医药有限公司,国药准字S10830001)总量100~125 mg/kg,分4 d静脉滴注,-10 d~-7 d;白消安(Bu 美国DSM pharmaceuticals,Inc,国药准字J20040107)0.8~1.2 mg/(kg/次)静脉滴注,每6 h 1次,共3 d,-8 d~-6 d;氟达拉滨(Flu重庆莱美药业股份有限公司,国药准字H20059418)40 mg/m2静脉滴注,共5 d,-7 d~-3 d;环磷酰胺(CY 江苏恒瑞医药股份有限公司,国药准字H32020857)40 mg/(kg·d)静脉滴注,共4 d,-6 d~-3 d。预防GVHD方案为-1 d开始予环孢素(杭州中美华东制药有限公司,国药准字H10960122)口服,监测药物浓度,谷浓度100~200 ng/mL,吗替麦考酚酯(MMF上海罗氏制药有限公司,国药准字H20031240)300 mg/(m2/次)口服,每12 h 1次,+1 d~+30 d;甲氨蝶呤(广东岭南制药有限公司,国药准字H20054692)15 mg/m2静脉滴注第1天,10 mg/m2静脉滴注第3天,第6天。输注干细胞数量(TNC)(4.50~8.70)×107/kg,CD34+(2.49~3.54)×105/kg。HSCT后1个月进行外周血嵌合状态及白细胞酶活性检测,采用短串联重复序列方法监测植入状态,当供者细胞比例≥95%为完全嵌合,酶学标本送检北京中科医学检验实验室。对5例患儿定期进行常规随访,随访时间8~27个月,末次随访时间为2023年5月30日。
2 结 果
2.1基本资料 5例可疑MPS患儿中男5例,女0例,所有患儿最初就诊医院的主诉均包括智力发育落后。确诊年龄3岁11个月~7岁5个月。其中MPSⅢA型4例, 其外周白细胞SGHS活性为2.2~5.8 nmol/(g·h)[参考:119.6~494.4 nmol/(g·h)];MPSⅢC型1例,外周白细胞HGSNAT活性为7.1 nmol/(g·h)[参考:276.2~660.5 nmol/(g·h)]。1例患儿临床表现包括躁动不安、面容粗糙、关节僵硬、进行性智力低下、语言发育落后、睡眠障碍;MPSⅢA型4例患儿有听力受损,部分患儿有反复呼吸道感染、运动发育落后、骨骼畸形、肝脾肿大、攻击行为等。见表1。
2.2基因变异分析 基因检测显示MPSⅢA型4例患儿SGSH基因均存在复合杂合变异,分别为c.863C>T(p.Pro2881Ile)和外显子1-8杂合缺失、c.703G>A(p.Asp235Asn)和c.1235T>C(p.Leu235Pro)、c.1129G>A(p.Arg377Cys)和c.1063G>T(p.Glu355Lys)、c.703G>A(p.Asp235Asn)和c.97G>A (p. Gly33Arg)。上述变异中的外显子1-8杂合缺失和c.1235T>C属于新发变异,未见文献报道,其余变异均有相关文献报道,根据ACMG评级,上述8个变异为可能致病性或致病性,见表2。MPSⅢC型1例基因检测显示HGSNAT基因存在复合杂合变异,分别为c.819T>G(p.Asn273Lys)和c.1277G>A (p.Gly426Glu),两个变异ACMG评级均为致病性变异,其中c.819T>G已有相关文献报道,c.1277G>A变异尚未见文献报道,属于新发现的致病性变异,见表2。
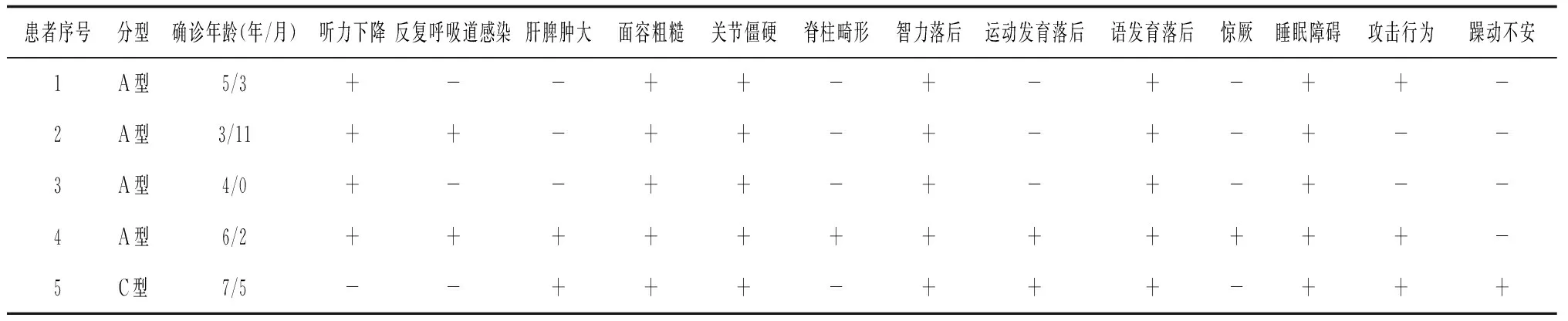
表1 5例MPSⅢ型患儿临床表现
2.3HSCT治疗效果及不良反应 经过脐带血HSCT治疗,5例患儿均获得顺利植入,+30 d查外周血嵌合均到达完全嵌合,+30 d查外周血白细胞酶活性均达到正常水平。随访8~27个月,5例患儿目前均存活,智力水平稳定,较移植前未见减退。移植后1个月酶值恢复正常,关节僵硬症状好转;移植后6个月,睡眠情况、躁动情绪、呼吸阻塞、呼吸道感染症状好转;移植12个月后,酶值稳定,身高、生长发育、骨骼异常等改善。见表3。
3 讨 论
MPS分为MPSⅠ型(Hurler综合征)、MPSⅡ型(Hunter综合征)、MPSⅢ型(Sanfilippo综合征)、MPSⅣ型(Morquio综合征)、MPSⅥ型(Maroteaux-Lamys综合征)、MPSⅦ型(Sly综合征)以及MPSⅨ型(Natowicz综合征)[1]。在亚洲,大多数MPS患儿属于MPSⅡ型,1984—2012年中国的一项MPS研究显示,506例MPS患儿中MPSⅡ型占所有MPS病例的近50%,MPSⅠ型约占13.7%,MPSⅢ型约占7.9%,MPSⅣ型约占24%,MPSⅥ型约占2.6%[16-17]。
MPSⅢ型中4个亚型特征分别为缺乏SGHS、NAGLU、HGSNAT和GNS,编码上述4种酶的基因分别是SGSH、NAGLU、HGSNAT和GNS[1,5]。由于HS在神经元发育中起着关键作用[18],因此可以解释MPSⅢ型患儿的神经病理学。通常MPSⅢ型患儿临床特征包括进行性特发性发育迟缓、认知能力下降、多动和睡眠障碍,当然不同亚型的临床特征各不相同,且患儿之间存在异质性[1,5]。
SGSH位于17q25.3,共14个外显子,编码502个氨基酸长度的蛋白质-硫酸乙酰肝素硫酸酯酶,该基因的突变是产生MPSⅢA型的病因。根据人类基因组数据库(HGMD数据库:http://www.hgmd.org),已有161种变异与MPSⅢA型有关,其中错义突变最多,为106种,移码突变30种,无义突变13种,大片段缺失4种,剪切位点3种,其他突变5种。本研究的4例MPSⅢA型患儿基因突变以错义突变为主(7个),缺失1个,错义突变发生在6,7,8外显子上,其中c.1235T>C,p.Leu235Pro是首次报道。患者2 SGSH基因的Exon 1-8杂合缺失也是首次报道。

表2 5例MPSⅢ患儿基因突变情况
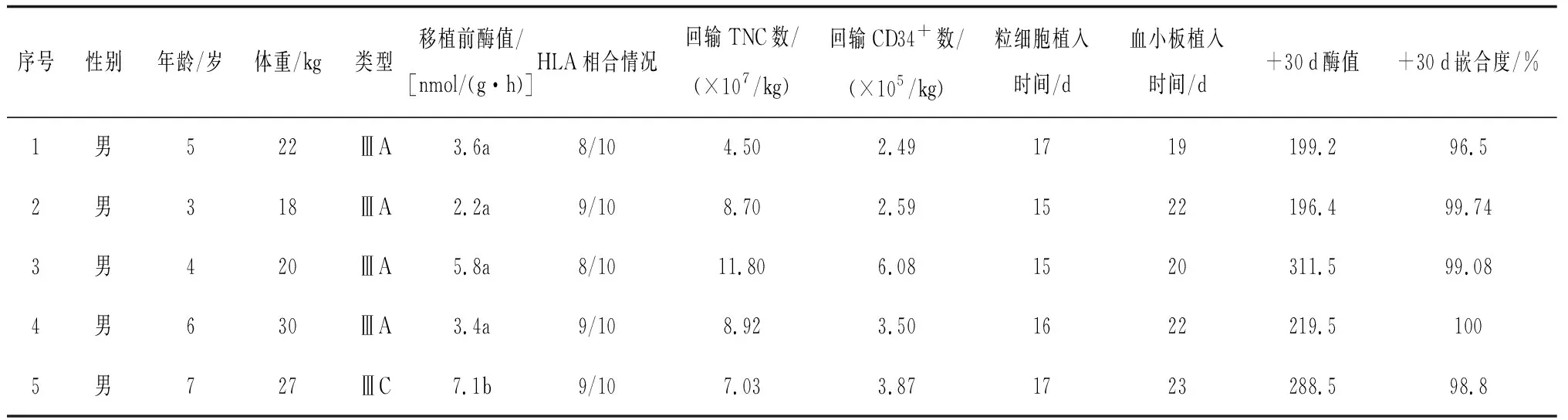
表3 5例MPSⅢ患儿造血干细胞移植治疗结果
HGSNAT位于8p11.21-p11.1,共20个外显子,编码663个氨基酸长度的蛋白质-乙酰CoA-氨基葡糖-N-乙酰转移酶,该基因的突变是产生MPSⅢC型的病因。根据HGMD数据库(http://www.hgmd.org),已有90种变异与MPSⅢC型有关,其中错义突变最多为37种,剪切位点16种,无义突变12种,移码突变11种,大片段缺失8种,同义突变1种。本研究的1例MPSⅢC型患儿HGSNAT基因发生2个错义突变(Exon 8和13),其中c.1277G>A,p.Gly426Glu是首次报道。
MPSⅢ患儿常见的初始症状包括发育迟缓、言语迟缓、反复耳鼻喉感染,以及某些患儿会发生腹泻或听力损失[19]。初始发育迟缓年龄1~4岁,经常伴有反复发作的耳部感染。在早期,患儿可能被误诊为特发性发育迟缓、注意力缺陷/多动障碍或自闭症谱系障碍,许多患儿随后发展为癫痫发作障碍。睡眠障碍很常见,会导致睡眠延迟、夜间频繁醒来和白天睡眠[20]。本研究中5例患儿均有智力、言语发育迟缓,其中3例患儿曾进行康复训练治疗,但均无改善。5例患儿均有不同程度的睡眠障碍,进行HSCT 6个月后均有不同程度的改善。
目前MPSⅠ型、MPSⅡ型国内外应用ERT治疗疗效已得到肯定,国内面临的问题是价格昂贵,而MPSⅢ型目前仍未有获批的酶替代药物,且大分子酶替代药物难以通过血脑屏障,所以中枢神经系统改善更为困难。检索国内文献,未找到HSCT治疗MPSⅢ型的报道,国外学者曾有报道MPSⅢ型患儿HSCT后均未提示神经系统症状有改善[21],故MPS的国内外指南和专家共识中,均未将MPSⅢ型的HSCT纳入其中。本研究中5例MPSⅢ型患儿经HSCT治疗后,患儿智力、语言等神经系统稳定,未出现继续减退现象,而睡眠、情绪方面大多已有改善,关节僵硬、感染情况改善较明显。但目前国内HSCT病例数少、随访时间短,HSCT用于MPSⅢ型患儿的治疗效果有待更大样本和长时间随访获得。
利益冲突:所有作者均声明不存在利益冲突。
猜你喜欢
广东药科大学学报(2023年5期)2023-12-30 00:08:39
分子催化(2022年1期)2022-11-02 07:11:08
时代报告·奔流(2022年1期)2022-04-29 04:10:56
昆明医科大学学报(2022年3期)2022-04-19 13:59:42
中国卫生(2016年4期)2016-11-12 13:24:10
中南医学科学杂志(2015年4期)2015-12-28 00:55:15
云南中医学院学报(2015年2期)2015-07-31 18:11:59
化学反应工程与工艺(2015年1期)2015-04-16 03:06:16
中国卫生(2014年4期)2014-12-06 05:57:06
中国塑料(2014年4期)2014-10-17 03:00:50
