
天然产物3′-prenylgenistein 的全合成
2023-12-09 08:17:14赵艳敏李红俊谢一民
北京化工大学学报(自然科学版) 2023年5期
赵艳敏 李红俊 谢一民 李 森
(1.银川能源学院化学与生物工程学院, 银川 750105;2.宁夏计量质量检验检测研究院, 银川 750411)
引 言
异黄酮类化合物广泛存在于植物中,具有多种生理及药理活性[1-5],近年的研究表明异黄酮类化合物在防治新型冠状病毒肺炎方面具有潜在的应用价值[6]。 3′-prenylgenistein 是一类新型的异戊烯基异黄酮,是由美国俄亥俄州立大学植物病理学家Cheng 等[7]从大豆中分离出来的,具体而言是由大豆疫霉菌病原体细胞壁中具有防御作用的葡聚糖激发子诱导而来。 该化合物对被高度表征的病原体衍生的葡聚糖激发子具有诱导积累作用,同时表现出良好的抑菌活性[7],而异戊烯基的存在使这种活性显著增强[8]。
目前,从天然产物中分离3′-prenylgenistein 存在分离困难、产率低、成本高等缺点,因此开展该异黄酮的全合成研究不仅具有重要的理论意义,而且具有潜在的实用价值。 人们对黄酮类化合物的合成进行了较多研究[9-13],但是对于3′-prenylgenistein全合成的研究未见报道。 本文以廉价的4-羟基苯甲醛和2,4,6-三羟基苯乙酮为原料,通过异戊烯基化、羟基保护、羟醛缩合、环化以及黄烷酮到异黄酮的重排等反应,完成了天然产物3′-prenylgenistein的全合成。
1 实验部分
1.1 实验材料与仪器
4-羟基苯甲醛、2,4,6-三羟基苯乙酮、乙酸乙酯、石油醚、二氯甲烷、甲醇、无水乙醇,分析纯,上海泰坦科技股份有限公司;GF254 硅胶,200 ~300 目(38 ~74 μm),青岛海洋化工厂。
AM-400 型核磁共振仪(NMR),溶剂为CDCl3,内标为四甲基甲硅烷(TMS),Bruker 公司;FTIR-8430S 型红外光谱仪(IR),日本岛津公司;HP-5988型电子离子化质谱仪(EI-MS),惠普公司。
1.2 实验方法
3′-prenylgenistein 的合成路线如图1 所示。 所有反应均用薄层色谱法(TLC)跟踪,采用柱色谱法纯化产品。
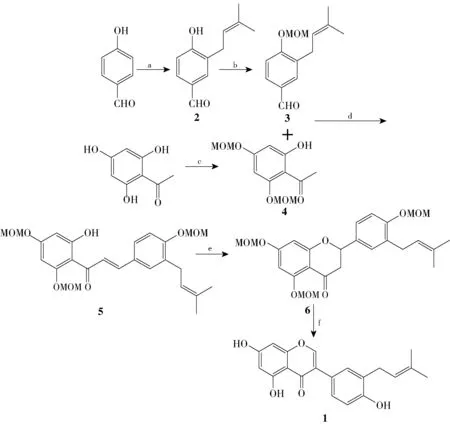
图1 3′-prenylgenistein 的合成路线Fig.1 Synthetic route of 3′-prenylgenistein
1.2.1 3-异戊烯基-4-羟基苯甲醛(2)的合成[14]
将氢氧化钾(1.12 g, 20 mmol)的水(20 mL)溶液冷却至0 ℃,磁力搅拌下加入4-羟基苯甲醛(1.22 g,10 mmol),待4-羟基苯甲醛全部溶解后,用注射器逐滴滴加异戊烯基溴(2.4 mL, 20 mmol),滴加完毕后继续在0 ℃下搅拌1 h,然后自然升至室温,避光继续搅拌10 h,用2 mol/L 的HCl 调至pH <4,用乙酸乙酯萃取(3 ×20 mL),合并有机相,依次用水和饱和NaCl 溶液洗涤,然后用MgSO4干燥。 过滤除去干燥剂,减压浓缩,通过硅胶柱层析分离(V(乙酸乙酯)∶V(石油醚) =1∶8),得到448 mg 无色油状物(化合物2),回收原料389 mg, 产率34.6%。
1H NMR(CDCl3, 400 MHz),δ: 9.88(1H, s,CHO),7.65(1H, d,J=7.2 Hz, ArH), 6.90(1H,d,J=8.0 Hz, ArH), 6.58(1H, s, ArH), 5.36(1H, t,J=7.6 Hz,CH= ), 3.45(2H, d,J=7.6 Hz, CH2),1.76,1.78(each 3H, each s,2 ×CH3);IR(KBr),vmax: 3 340, 2 924, 1 375, 1 363 cm-1;EI-MS(70 eV),m/z(%)[M]+: 190(2.2), 147(100),135(32),107(42),91(64),43(66)。
1.2.2 3-异戊烯基-4-甲氧甲氧基-苯甲醛(3)的合成
将1.2.1 节纯化后的化合物2(190 mg, 1.0 mmol)溶于干燥的丙酮(10.0 mL)中,快速搅拌下加入无水K2CO3(166 mg, 1.2 mmol),待溶解后缓慢滴加氯甲基甲基醚(96 mg, 1.2 mmol),滴加完毕后加热回流0.5 h,冷却至室温,萃取、干燥、减压浓缩,通过硅胶柱层析分离(V(石油醚)∶V(乙酸乙酯) =16∶1),得到213 mg 化合物3,产率96.2%。
1H NMR (CDCl3, 400 MHz),δ: 9.87(1H, s,CHO),7.89(1H, s, OH),7.67(2H, m, H-2 and H-6), 7.16(1H, d,J=8.0 Hz, H -5), 5.34(1H,t,J=7.0 Hz,CH= ),5.29(2H,s,OCH2O),3.49(3H, s,J=7.0 Hz, OCH3),3.38(2H, d,J=7.6 Hz, CH2), 1.75(3H, s, CH3), 1.73(3H, s,CH3); EI-MS(70 eV),m/z(%)[M]+:222(19),207 (100),115(37),91(13),51(16)。
1.2.3 2-羟基-4,6-二甲氧甲氧基苯乙酮(4)的合成
将2,4,6-三羟基苯乙酮(36.0 mg, 2.0 mmol)溶于20.0 mL 干燥的丙酮中,按照1.2.2 节的操作步骤最终得到471.0 mg 淡黄色油状物(化合物4),产率91.0%。
1H NMR (CDCl3,400 MHz),δ:13.56(s,1H,OH),6.47(d,J=2.4 Hz,1H,ArH),6.13(d,J=2.4 Hz, 1H, ArH), 5.45(s, 2H, OCH2O), 5.23(s,2H, OCH2O), 3.36(s, 3H, OCH3), 3.24(s,3H, OCH3), 2.78(s, 3H, COCH3); IR(KBr),vmax: 2 950, 2 945, 1 665, 1 440, 1 350, 1 205,1 200, 1 150, 1 060 cm-1; EI -MS(70 eV),m/z(%)[M]+: 256(16), 211(7), 182(12), 45(100)。
1.2.4 3′-异戊烯基-2-羟基-4,6,4′-三甲氧甲氧基查尔酮(5)的合成[15-16]
在氮气保护下,将KOH(2.8 g, 50.0 mmol)、乙醇(3 mL)、H2O(2 mL)的混合液缓慢滴加到化合物4(256.0 mg,1.0 mmol)和化合物3(280.8 mg, 1.2 mmol)的乙醇(2.0 mL)溶液中,滴加完毕后冷却至0 ℃并继续在该温度下反应1 h,然后在室温下搅拌24 h,加入冰水淬灭反应,用3 mol/L 的HCl 调节pH <2,用CH2Cl2萃取(3 ×5 mL),合并有机相,依次用水和饱和NaCl 溶液洗涤,干燥,减压浓缩,通过硅胶柱层析分离(V(乙酸乙酯)∶V(石油醚) =1∶10),得到378.2 mg 淡黄色固体(化合物5),产率80.1%。
1H NMR(CDCl3, 400 MHz),δ: 13.37(s, 1H,OH),7.85(d,J=8.4 Hz, 1H, H -6′), 7.83(d,J=15.2 Hz, 1H, H - β), 7.46(d,J=15.2 Hz,1H, H- α), 7.39(s, 3H, H -2,4 and 6), 6.74(d,J=2.4 Hz, 1H, H -5′), 6.60(dd,J=8.8,2.4 Hz,1H, H-5′),5.29(m,2H, H-2″),5.23,5.12, 5.04 (each s, each 2H, OCH2O), 3.77,3.52,3.49(each s, each 3H, OCH3),3.42(d,J=7.2 Hz, 2H, H - 1″), 1.77, 1.78 (each s, each 3H, H - 4″, 5″); IR(KBr),vmax: 2 940, 2 910,1 676, 1 515, 1 495, 1 460, 1 305, 1 260, 1 156 cm-1; EI-MS(70 eV),m/z(%)[M]+: 472(22),441(7),427(2),45(76),43(100)。
1.2.5 3′-异戊烯基-5,7,4′-三甲氧甲氧基黄烷酮(6)的合成
将无水乙酸钠加入到化合物5 (94.4 mg,0.2 mmol)的甲醇(1 mL)溶液中,然后加入两滴水,加热回流10 h,冷却至室温,加入少量水,用CH2Cl2萃取(3 ×10 mL),合并有机相,依次用水和饱和NaCl 溶液洗涤,干燥,过滤除去干燥剂,减压浓缩,通过硅胶柱层析分离(V(石油醚)∶V(乙酸乙酯) =6∶1),得到76.2 mg 淡黄色油状物(化合物6),回收原料10.8 mg,产率92.2%。
1H NMR(CDCl3, 400 MHz),δ: 7.05(dd,J=2.4,2.8 Hz, 1H, H -6′), 7.00(d,J=2.4 Hz,1H, H -2′), 6.86(1H, d,J=7.6 Hz, H -5′),6.44(1H, d,J=3.2 Hz, ArH), 6.39(1H, d,J=2.0 Hz, ArH), 5.32(1H, dd,J=3.2, 14.0 Hz, H-2), 5.17, 5.19, 5.12(each 2H, s, 3 × OCH2O),3.47, 3.54, 3.59(each 3H, s, 3 × OCH3), 3.02(1H, dd,J=13.2, 16.4 Hz, H -3ax), 2.75(1H,dd,J=2.8,16.4 Hz, H -3eq), 5.62(1H, d,J=7.2 Hz,H-2″),3.94(2H,d,J=8.4 Hz,H-1″),1.79(3H, s, H -3″), 1.76(3H, s, H -3″); IR(KBr),vmax: 2 825, 1 694, 1 628, 1 360, 1 149 cm-1; EI-MS(70 eV),m/z(%)[M]+: 472(1),457(8),427(12),271(38),45(100)。
1.2.6 3′-prenylgenistein(1)的合成[3]
将化合物6(472 mg, 1.0 mmol)溶于三甲基甲酸酯(15 mL),然后逐滴加入浓硫酸(20 μL),滴加完毕后,在常温下搅拌25 min,然后逐滴滴加BTI(645 mg,1.5 mol)的三甲基甲酸酯(3 mL)的混合液,滴加完毕后继续在常温下搅拌24 h,减压蒸除溶剂后加入20 mL 蒸馏水,继续在常温下搅拌2 h,用乙酸乙酯萃取(3 ×30 mL),通过无水MgSO4干燥,减压蒸除溶剂,加入20 mL 无水甲醇,滴加6 mol/L的HCl (0.5 mL, 6.0 mmol),滴加完毕后加热回流2 h,加入25 mL 蒸馏水,用乙酸乙酯萃取(3 ×30 mL),合并有机相,依次用水和饱和NaCl 溶液洗涤,通过无水MgSO4干燥,过滤除去干燥剂,减压蒸除溶剂,将剩余物通过硅胶柱层析分离(V(乙酸乙酯)∶V(己烷) =3∶1 ~2∶3),得到201 mg 淡黄色固体异黄酮(化合物1),产率60.0%。
1H NMR(CDCl3, 400 MHz),δ: 8.22(1H, s,—OH), 7.96 (1H, d,J= 8.0 Hz, ArH), 7.62(1H, d,J=8.4 Hz, ArH),7.36(1H, dd,J=8.0,8.4 Hz, ArH), 7.30(1H, d,J=2.4 Hz, ArH),7.00(1H, d,J=7.6 Hz, ArH),5.32(1H, tm,J=7.2 Hz,H-2″),3.35(2H,d,J=7.6 Hz,H-1″),1.72(3H, s, H - 3″), 1.72(3H, s, H - 3″);13C NMR(100 MHz, acetone-d6),δ: 178.9, 158.6,154.5, 151.8, 147.8, 134.0, 133.1, 131.2,130.9, 128.9, 125.2, 125.0, 123.6, 118.9,117.2,115.3,111.2,56.0,29.8,25.8,17.4; IR(KBr),vmax: 2 678, 1 786, 1 456, 1 360, 1 321 cm-1; EI-MS(70 eV),m/z(%)[M]+: 338(4),321(5),291(2),274(32),171(37),45(100)。
2 结果与讨论
4-羟基苯甲醛在强碱条件下与异戊烯基溴反应,以34.6%的收率得到主产物为C-异戊烯基化的化合物2;化合物2 在K2CO3的作用下与氯甲基甲基醚反应,以96.2%的收率得到化合物3;同时,以2,4,6-三羟基苯乙酮为起始原料在弱碱性条件下与氯甲基甲基醚反应,以91.0%的产率得到化合物4,化合物4 的2 位羟基不与氯甲基甲基醚反应,主要原因是2 位上的羟基和1 位上的羰基形成分子内氢键,降低了其反应活性;化合物3 和4 在氮气保护下在强碱作用下反应,以80.1%的产率得到查尔酮类化合物5;化合物5 在无水乙酸钠的作用下关环,以92.2%的产率得到黄烷酮6;化合物6 在BTI 的作用下发生黄烷酮到异黄酮的重排反应,以60.0%的收率合成了最终的天然产物3′-prenylgenistein,其波谱数据与文献[7]报道的结果一致。 经上述反应,3′-prenylgenistein 的总产率为13.4%。
在以上3′-prenylgenistein 的合成方法中,由黄烷酮合成异黄酮是合成的关键步骤,在多功能氧化剂BTI 的作用下黄烷酮重排合成异黄酮。 在以往由黄烷酮重排合成异黄酮的方法中,分为两步得到最终产物,第一步是黄烷酮先脱去MOM 保护(收率92%),第二步是脱去保护后再重排生成异黄酮(收率57%),这两步的总收率为52%。 而本实验实现了重排与脱保护的一锅法合成异黄酮,该方法简单快捷,收率相对较高(60%),为异黄酮的合成提供了新思路。
3 结论
本文以廉价的4-羟基苯甲醛和2,4,6-三羟基苯乙酮为起始原料,通过异戊烯基化、羟基保护、羟醛缩合、环化以及黄烷酮到异黄酮的重排等步骤,以13.4%的总产率完成了天然产物3′-prenylgenistein的全合成。
猜你喜欢
中国药业(2020年7期)2020-04-11 05:48:08
消费导刊(2019年14期)2019-08-21 01:00:51
消费导刊(2019年27期)2019-07-22 09:12:22
天然产物研究与开发(2018年10期)2018-11-06 07:43:52
天然产物研究与开发(2018年10期)2018-11-06 07:43:42
中国农业科学(2017年5期)2017-03-22 06:47:52
合成化学(2015年2期)2016-01-17 09:03:25
中国医学科学院学报(2015年5期)2015-03-01 04:03:35
合成化学(2014年2期)2014-06-23 16:22:14
天然产物研究与开发(2014年6期)2014-04-27 14:16:00
