
用X射线衍射法分析多晶型药物晶型一致性
2023-12-05 07:19:04王帅陈永康张笑旻王勇上海市计量测试技术研究院
上海计量测试 2023年5期
王帅 陈永康 张笑旻 王勇 / 上海市计量测试技术研究院
0 引言
药物多晶型是指固体药物的分子排列规律不同、分子构型或构象存在差异,从而表现出不同的存在形态。Woehler 和Liebig 在1832 年发现苯甲酰胺化合物存在两种不同的晶型[1],自此越来越多的研究者发现多数药物可以形成两种或多种晶型物质。近年来,药物晶型的研究越来越受到重视。同一药物的不同晶型之间由于晶格不同,质点间的相互作用力和结合能力也不同,其理化性质会有所改变,从而导致不同晶型药物体内生物利用度、有效性的差异,有的甚至会引发毒性[2]。医药研发领域中多晶型现象势必影响新药的生物利用度或仿制药一致性评价中的生物等效性,所以药物多晶型现象是影响药品质量和临床疗效的重要因素之一。因此,对存在多晶型的药物进行研发及审批时,必须对药物晶型进行一致性确认。
X 射线衍射(X-ray diffraction,XRD)技术是研究物质晶型的主要手段之一。XRD 法的原理是使用单色X 射线照射样品,不同晶面会产生不同的衍射图谱,采用晶面间距dhkl(Å)或掠射角θ(°) 和衍射峰强度来定量表示衍射图谱[3]。XRD 法作为一种非破坏性的晶型分析方法,可区分混合物和化合物,物质的晶态和非晶态,也可用于鉴别晶体的种类和结构,测试晶胞参数等。XRD 法作为一种常用的分析技术,具备快速、准确、非破坏性等优点,因此,XRD 法是判断药物晶型的主要检测手段。本研究使用XRD 法对不同批次样品进行晶型一致性确认,并对所用方法进行了方法学研究,确保方法的准确可靠。
1 材料和方法
1.1 材料
1.1.1 仪器设备
X 射线衍射仪:Smart Lab(Rigaku 公司);红外光谱仪:Spectrum 3(Perkin Elmer 公司);差示扫描量热仪:Diamond DSC(Perkin Elmer 公司)。
1.1.2 样品试剂
样品A(批号:1,2,3);样品A 标准品。
1.2 方法
1.2.1 一致性评价方法
本研究选用具有晶型结构的多个批次药物样品及标准品,使用X 射线衍射仪获取其衍射图谱。通过分析主要特征峰相对强度及2θ值,判断晶型一致性。XRD 法的条件如下:取适量的固体样品均匀平铺于样品盘上,使用玻璃片压平样品,如有颗粒,轻压使其呈粉末状。保持固体样品表面平整且与样品盘表面在同一水平面上。设置波长Cu Kα-1.540 6(Å);扫描角度: 3°~42°;扫描步长:0.02°;扫描速度: 0.2 s/步,电压40 V,电流40 mA。通过对样品和标准品的衍射图谱进行比较和分析,评估不同批次样品与标准品的晶型差异,确定晶型的一致性。并使用红外光谱仪和差示扫描量热仪对样品及标准品进行测试,以佐证晶型结果。
1.2.2 方法确认
参照《中华人民共和国药典(2020 年版)》中9101 分析方法验证指导原则及9015 药品晶型研究及晶型质量控制指导原则[4-5],考察方法的精密度(重复性和中间精密度)、专属性、耐用性。具体方法如下:1)重复性:按照分析方法,确定标准品的特征衍射峰。分别检测6 个标准品及样品,并计算标准品及样品的主要特征峰的2θ值相对标准偏差(RSD),可接受标准:RSD≤5%。2)中间精密度:在不同时间段由不同人员进行方法重复性研究,可接受标准:RSD≤5%。3)专属性:在相同测试条件下,同一晶型的样品与标准品的主要特征峰2θ值的偏差均应小于0.2°。4) 耐用性:改变仪器电压参数,计算结果之间的偏差,标准品及样品的2θ值偏差均应小于0.2°。
2 结果
2.1 晶型一致性试验
本研究选用样品A 作为研究对象,其三个批次样品及标准品XRD 图谱如图1 所示。由于衍射峰强度顺序及峰强度易受到晶体粒径或形貌排列导致的择优取向影响,因此,不作为一致性判别依据[6]。衍射图谱中的衍射峰数量相同,2θ值衍射峰位置误差范围在±0.2°内,衍射峰的强弱顺序一致,说明样品的晶型一致。

图1 样品A 三批次及标准品XRD 图谱
使用红外光谱仪和差示扫描量热仪(DSC)对样品A 三个批次及标准品进行测试,红外光谱如图2 所示,DSC 曲线图如图3 所示。

图2 样品A 三批次及标准品红外光谱图
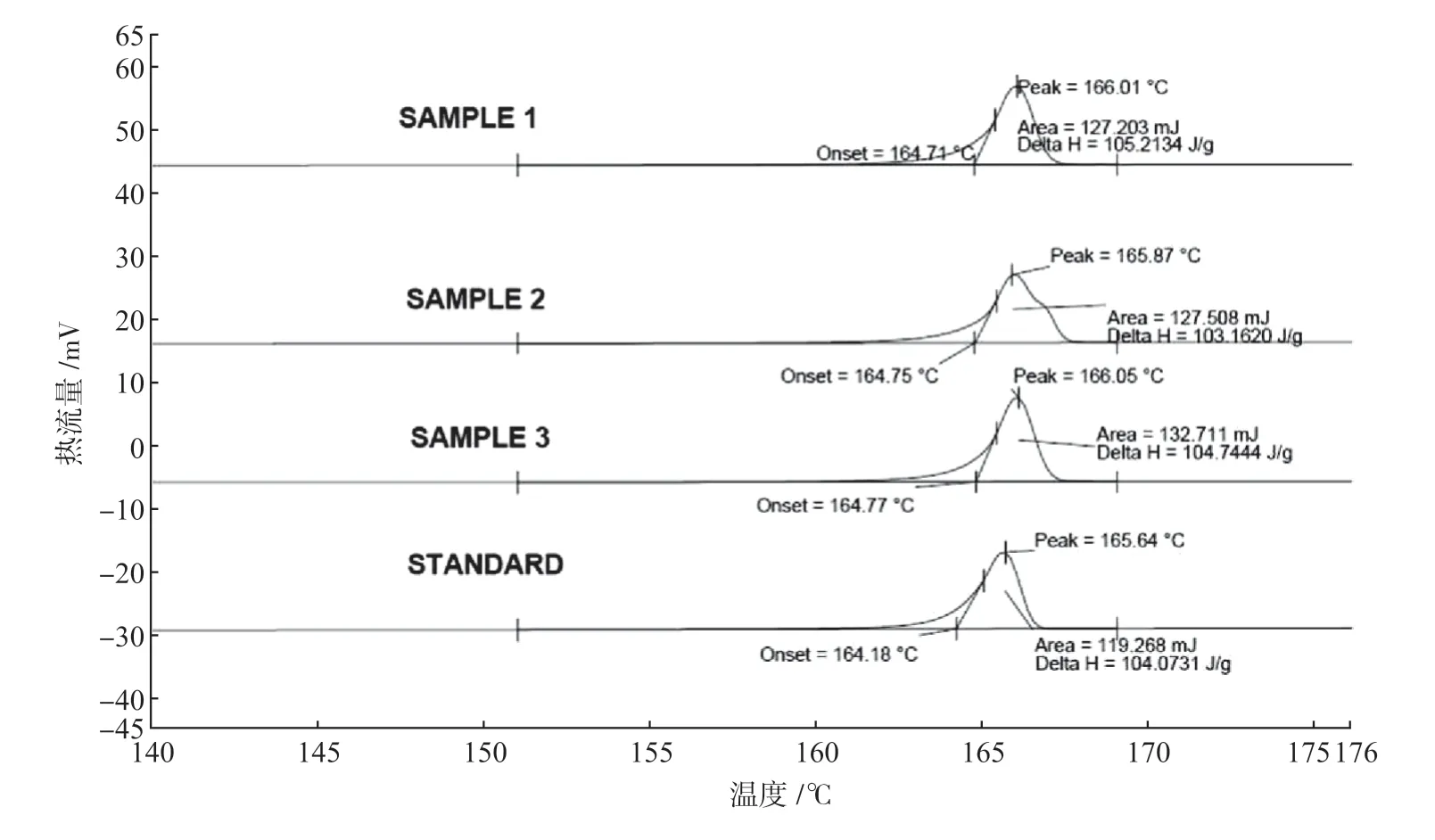
图3 样品A 三批次及标准品DSC 曲线
不同晶型分子的化学键键长、键角不同,其振动-转动跃迁能级不同,在红外光谱中的表现为吸收带频率、峰形、峰位、峰强度等出现差异。图2中样品A 三批次及标准品的结果基本一致,可以作为批次样品与标准品晶型一致性判断的佐证。
不同晶型物质在升温或冷却过程中的吸、放热也会有差异。差示扫描量热仪通过不断加热或降温,测量样品与惰性参比物之间的能量差随温度的变化,并通过测得的热分析曲线来判断药物晶型的异同。该方法容易受升温速率及仪器本身状态影响,适合用于热峰值存在较大差异的晶体鉴别。图3 中样品A三批次及标准品的起始放热温度、放热峰值、热焓值基本一致,可作为批次样品与标准品晶型一致性判断的佐证。
2.2 方法学验证试验
2.2.1 重复性
6 个样品及6 个标准品的主要特征峰(11 个)的2θ值相对标准偏差(RSD)结果如表1 所示。

表1 重复性样品及标准品主要特征峰2θ 值RSD
样品的11 个主要特征峰2θ值的RSD在0.06%~0.21%之间,标准品的11 个主要特征峰2θ值RSD在0.03%~0.12%之间,均符合可接受标准(RSD≤5%),方法重复性良好。
2.2.2 中间精密度
在不同时间段由不同人员进行方法重复性研究,12 个样品及12 个标准品的主要特征峰的2θ值相对标准偏差结果如表2 所示。

表2 中间精密度样品及标准品主要特征峰2θ 值RSD
样品的11 个主要特征峰2θ值的RSD在0.05%~0.18% 之间,标准品的11 个主要特征峰2θ值RSD在0.06%~0.24%之间,均符合可接受标准(RSD≤5%),方法中间精密度良好。
2.2.3 专属性
在相同测试条件下,同一晶型的样品与标准品的主要特征峰2θ值及偏差如表3 所示。
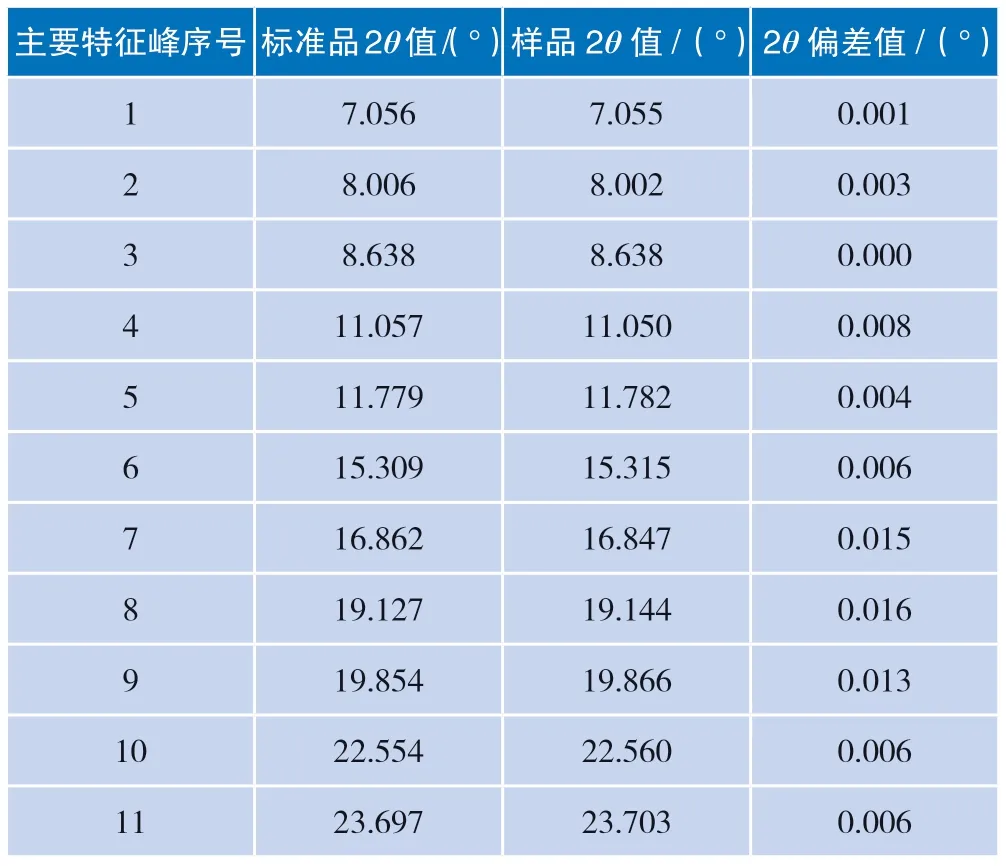
表3 样品与标准品的主要特征峰2θ 值及偏差
以上结果表明样品与标准品的11 个主要特征峰的2θ值偏差值最大为0.016,均小于0.2°,方法专属性良好。
2.2.4 耐用性
改变仪器电流参数为45 mA 后,对样品及标准品进行测试,比较不同测试条件下样品的2θ值偏差,其结果如表4 所示。
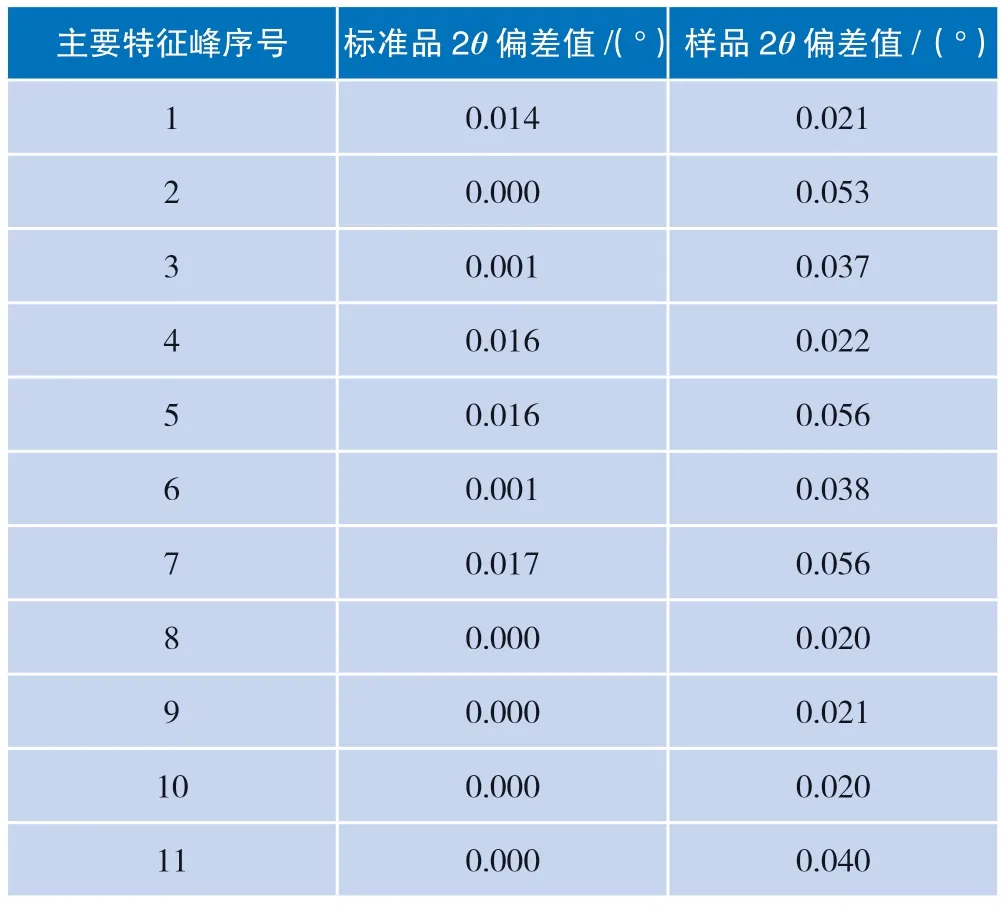
表4 不同电压下样品与标准品的主要特征峰2θ 值偏差
不同测试条件下的供试品11 个主要特征峰的2θ偏差值均小于0.2°;不同测试条件下的标准品11个主要特征峰的2θ偏差值均小于0.2°,均符合可接受标准(2θ值偏差应小于0.2°),表明方法耐用性良好。
3 结语
使用X 射线衍射法对多晶型药物进行晶型一致性研究,并对该方法进行方法学验证。结果表明该方法具有良好的精密度、专属性和耐用性。因此,X射线衍射法可以用于确定不同批次间药物样品与标准品的晶型差异,并评估晶型的一致性。
药物晶型在制剂过程如固体分散体、研磨、制粒、冷冻干燥及喷雾干燥时往往会发生晶型转变,影响制剂的质量[4]。使用XRD 法对药物晶型进行一致性判断,是一种简单有效的方法,也已被多国药典收录。但由于医药行业发展迅速,许多创新药没有完备的XRD 检测方法,且由于药物晶型粒度会引起择优取向,对衍射峰和相对强度产生影响,很可能导致同一晶型被误判为不同晶型[5]。因此,对多晶型药物一致性评价方法进行开发及方法学验证具有一定的必要性。在某些特殊情况下还应选择多种分析方法综合判断晶型一致性结果,以避免误判。
猜你喜欢
青岛科技大学学报(自然科学版)(2023年6期)2023-11-25 17:17:56
昆钢科技(2021年2期)2021-07-22 07:46:56
陶瓷学报(2020年2期)2020-10-27 02:16:14
World Journal of Clinical Cases(2020年17期)2020-09-18 08:03:24
检验医学与临床(2020年1期)2020-01-10 04:44:22
职工法律天地·上半月(2018年12期)2018-01-22 22:55:10
水利信息化(2017年4期)2017-09-15 12:01:21
应用海洋学学报(2015年2期)2015-11-22 07:36:40
中国塑料(2015年6期)2015-11-13 03:02:34
中国塑料(2015年8期)2015-10-14 01:10:48
