
14例木村病患者的临床特征及文献复习
2023-11-17 02:44:54张靖岚刘钰万鼎铭王萌
河南大学学报(医学版) 2023年5期
张靖岚,刘钰,万鼎铭,王萌
郑州大学第一附属医院 血液科,郑州 450052
木村病(Kimura's disease,KD)又名嗜酸性粒细胞增生性淋巴肉芽肿,是一种不明原因的慢性炎症性疾病,多表现为缓慢进展的无痛性浅表淋巴结肿大和(或)软组织包块,临床非常罕见,目前尚无标准的治疗方案。本文对2016年10月至2021年10月我科收治的14例木村病患者的病例资料进行回顾性分析并文献复习,报告如下。
1 对象与方法
收集2016年10月至2021年9月我科收治的经病理检查确诊为木村病的14例患者的病例资料。本组患者男性11例、女性3例;起病时年龄16~62岁,<20岁2例,20~40岁8例,>40岁4例;病程1~20 a,>5 a者有5例,>10 a者有4例。
从病史、临床表现、辅助检查等几个方面对以上患者的临床特征进行归纳总结,并对以上患者进行随访。
2 结果
2.1 临床表现
14例患者初发症状均为局部包块,直径2.0~4.0 cm,质韧,均无疼痛、压痛、压迫症状等不适。其中以头颈部肿块最多见,有13例,占所有病例的92.9%。14例患者中4例同时伴有腋窝或腹股沟淋巴结肿大。病灶可单发(7例,50.0%),亦可多发(7例,50.0%)。病变部位主要涉及颈部(6 例,42.9%)、右侧下颌部(4 例,28.6%)、耳后(3 例,21.4%)、腮腺 (2例,14.3%)、腋窝(3例,21.4%),腹股沟(2例,14.3%)。
10例患者起病时伴有皮肤瘙痒,其中1例患者出现背部散在红色皮疹、伴瘙痒、无脱屑。
2.2 辅助检查
14例患者外周血嗜酸性粒细胞比例均显著增高(17.6%~48.9%),均行血清IgE检测,IgE水平均高于正常(815.1~2337.0 IU/mL)。其中6例患者行骨穿检查,骨髓检查提示均示:骨髓增生活跃,粒系增生活跃,嗜酸性粒细胞比值偏高,余未见异常。14例患者检查肾功能均正常,其中2例出现肾病综合征,大量蛋白尿,尿蛋白定量3.0~5.2 g/24 h。
14例患者进行超声检查提示头颈部肿块或腋窝、腹股沟淋巴结肿大,均行肿物切除活检,病理结果显示:淋巴滤泡增生活跃,见大量嗜酸性粒细胞浸润,可伴有血管增生和纤维化。进一步行免疫组化检查支持木村病诊断。14例患者中2例患者出现胃肠道受累,行胃肠镜检查,镜下提示慢性胃炎,慢性结肠炎。胃镜下取组织活检示:黏膜固有层可见嗜酸性粒细胞浸润。2例肾脏受累,表现为肾病综合征,进一步行肾穿刺活检,1例病理提示为微小病变肾小球病,另1例提示为微小病变肾小球病伴轻度系膜增生性IgA肾病,见图1。
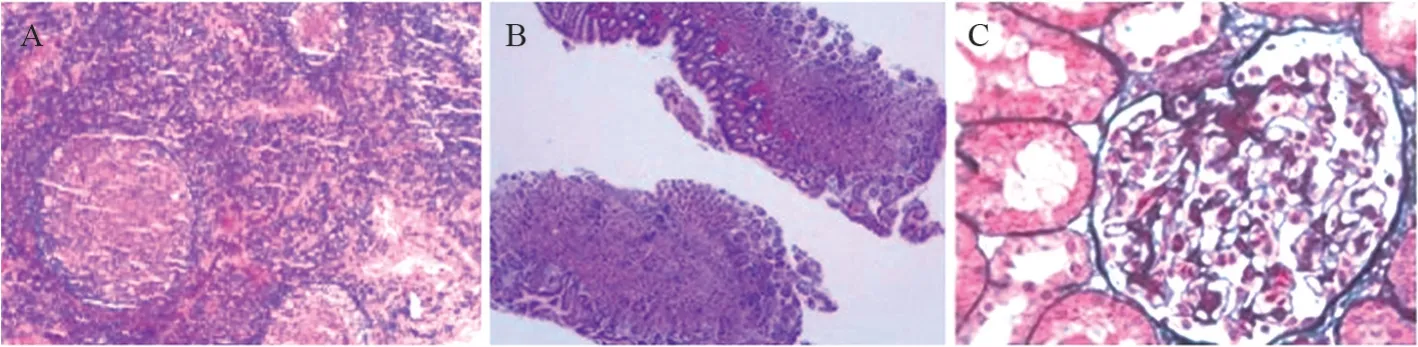
图1 KD患者的组织病理
2.3 治疗及转归
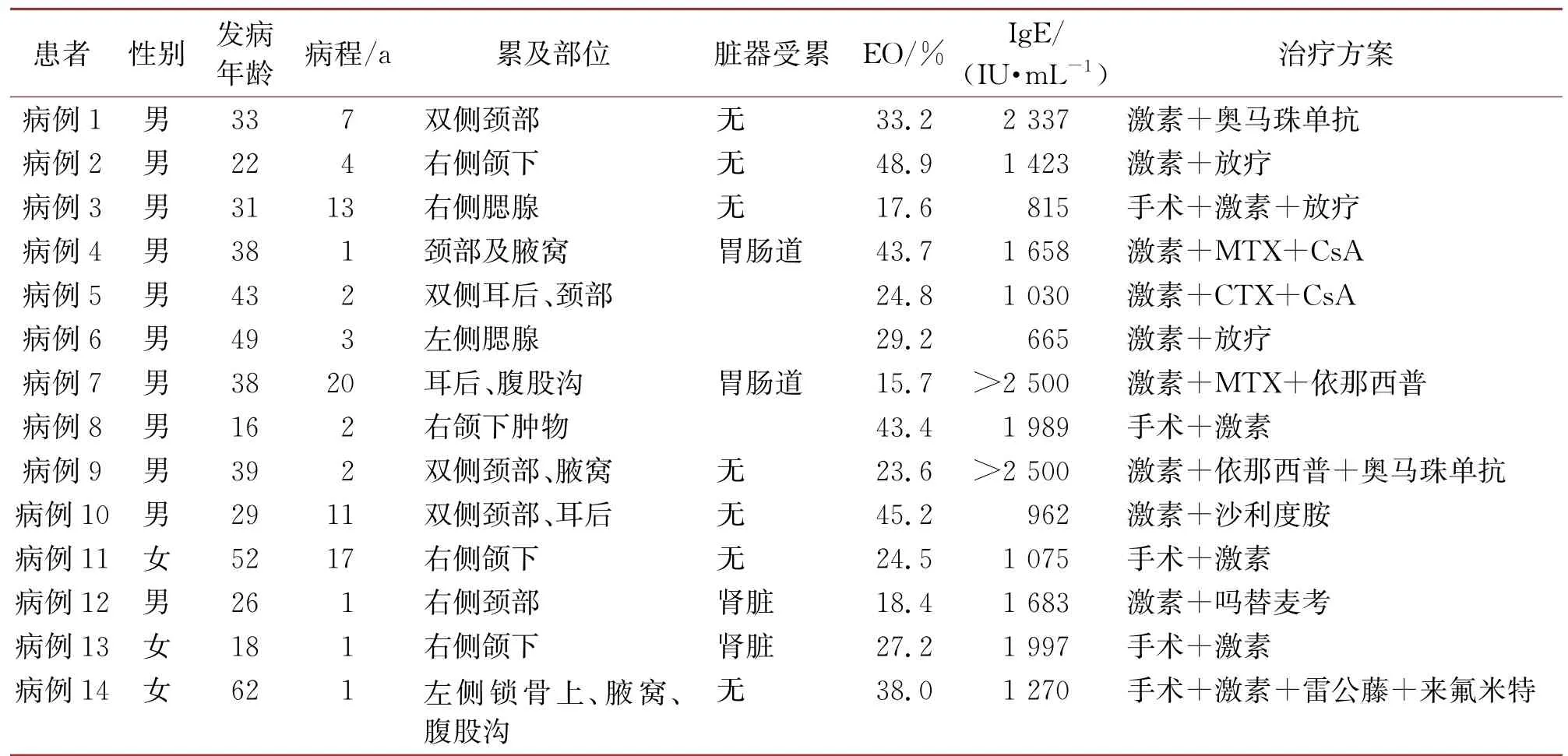
表1 14例KD患者的临床特征及治疗转归
确诊后5例患者行手术治疗,术后给予激素维持。其余9名患者未行手术,单纯给予激素治疗。给予激素治疗后疗效明显,肿块明显缩小,嗜酸性粒细胞及IgE水平显著下降。但11例(78.6%)患者在激素减量或停用后复发,其中3例加用放疗后未再复发。其余8例患者加用不同种类免疫抑制剂或奥马珠单抗(IgE单抗)等维持治疗。病例1曾加用奥马珠单抗治疗疗效不理想,后放弃治疗,目前仍双侧颈部淋巴结肿大,伴有严重皮肤瘙痒,患者处于焦虑状态。其余患者加用MTX、CsA、依那西普、吗替麦考酚酸酯等免疫抑制剂维持治疗至今。目前所有14例患者仍存活,病程最长者达20 a。
3 讨论
KD好发于亚洲青中年男性,病程较长且进展缓慢。目前其病因及发病机制尚不明确,可能与寄生虫、病毒、真菌感染,昆虫叮咬等引起的过敏反应有关,未知抗原刺激肥大细胞释放IgE,诱发Ⅰ型变态反应可能是其发病机制[1-2]。
KD为局部慢性炎症性病变,以进行性、无痛性增大的皮下肿块为主要表现,多见于头颈部腺体、淋巴结及软组织,腋窝、腹股沟、四肢、躯干及内脏等部位也可累及[2-3]。本研究中14例患者均表现为无痛性肿块,其中13例患者出现头颈部肿块,与既往文献报道一致。肿块表面皮肤多正常,可伴有瘙痒、皮疹等不适,本组10例患者伴有明显皮肤瘙痒,皮肤瘙痒考虑与淋巴细胞和嗜酸性粒细胞增多后释放的细胞因子及神经递质浸润感觉神经纤维有关[4]。
该病可累及多脏器,其中肾脏受累也较常见,12%~16%的KD患者可出现蛋白尿,其中60%~80%表现为肾病综合征[5-6]。本研究中14例患者中2例患者胃肠道受累,2例患者肾脏受累,均表现为肾病综合征,大量蛋白尿,但未出现肾功能衰竭,肾脏穿刺活检提示微小病变肾小球病,经激素及免疫抑制剂等治疗后,蛋白尿减轻。
外周血嗜酸性粒细胞增多及血清IgE 升高是KD的主要实验室特点[2,6-7]。本组14例患者IgE水平及嗜酸性粒细胞比例均显著增高。影像学在KD诊断上作用有限,病变组织的病理检查是诊断该病的金标准。典型的病理特点是显著的淋巴滤泡增生,周围大量淋巴细胞、嗜酸性粒细胞浸润,易形成嗜酸性微脓肿,可伴有血管增生和纤维化,免疫组化可见滤泡内以B淋巴细胞浸润为主,滤泡间以T淋巴细胞浸润为主,IgE在生发中心沉积[2,8-9]。部分患者在病理活检前常被误诊为淋巴瘤、血管淋巴样增生伴嗜酸性粒细胞增多症、Castelman病等。当患者出现无痛性肿块,尤其头颈部肿块,并伴有IgE和嗜酸性粒细胞显著增高时,应考虑KD,需及时对病变组织进行活检有助于明确诊断。
本组14例患者病程最长达20 a,病程>10 a者有4例。病例1拒绝维持治疗,随访患者主要表现为双侧颈部淋巴结肿大,严重皮肤瘙痒,未发现明显脏器受累。其余患者目前均病情稳定。提示KD属于良性病变,预后较好,可长期生存。但KD容易复发,目前尚无标准化的治疗方案,临床上采用的治疗方式包括手术切除、糖皮质激素、局部放射、细胞毒性药物及联合治疗等[2,6-7]。由于手术切除兼具诊断及治疗作用,对于孤立的、局限的病灶建议作为首选,糖皮质激素也是KD 常用的治疗手段,疗效显著,但单用激素治疗停药后易复发,目前推荐长期、小剂量维持治疗,有助于防治伴发的肾脏疾病[2,6-7]。文献报道[10]显示,放疗在KD治疗中具有非常高的价值,推荐剂量为26~30Gy,疗程为2~3周,本组3例患者在复发后行局部放疗,随访3~4 a未见复发,且未发现明显的放疗相关的毒副作用。近年来有文献报道[11],采用IgE抗体-奥马珠单抗(omalizumab)治疗KD有效,但病例1应用该药后疗效不明显。此外有研究[6,12-13]显示,环孢素A、沙利度胺、MTX、MMF等药物对木村病均有一定疗效,对于激素停药后复发或激素依赖患者可考虑联合CsA等免疫抑制剂治疗,但目前对于维持治疗的剂量及疗程仍需进一步探索。
综上所述,KD在临床上较为少见,对于出现无痛性皮下包块、高嗜酸性粒细胞、IgE显著增高的患者,应高度怀疑本病,尽早完善病理检查以明确诊断。KD的治疗效果因人而异,激素治疗效果显著但减量或停药后易复发,联合放疗或免疫抑制剂可减少复发。
猜你喜欢
中国药学药品知识仓库(2022年9期)2022-05-23 00:30:46
健康体检与管理(2022年2期)2022-04-15 01:33:37
中国临床医学影像杂志(2019年4期)2019-06-18 10:54:50
基层中医药(2018年1期)2018-03-01 07:36:21
中成药(2017年4期)2017-05-17 06:09:52
中华老年口腔医学杂志(2016年2期)2017-01-15 14:24:54
台声(2016年10期)2016-09-19 05:46:28
中国卫生标准管理(2015年14期)2015-01-27 02:24:27
恋爱婚姻家庭·养生版(2014年9期)2014-08-26 15:55:26
中华介入放射学电子杂志(2014年1期)2014-02-02 05:24:07
