
基于对比学习的蛋白质功能预测模型
2023-11-10 08:07:52孙旭,林劼
福建师范大学学报(自然科学版) 2023年6期
孙 旭,林 劼
(1.福建师范大学数学与统计学院,福建 福州 350117;2.福建师范大学计算机与网络空间安全学院,福建 福州 350117)
蛋白质在生命活动中承担着多种基本功能和作用,如代谢调控、基因表达调控、抵抗疾病等[1].蛋白质功能的发现有助于理解生物体内疾病与基因之间的内在机理和联系[2],进而研究开发出针对新型冠状病毒肺炎、艾滋病病毒或天花病毒等全球性传染病的新药.
到目前为止UniProt数据库中仅对不到1%的蛋白质进行了实验注释.对未知蛋白质进行实验注释代价高昂且耗时,并且由于未知蛋白质数量的不断增加也使得无法缩小对蛋白质进行功能注释的数量与未知蛋白质数量之间的差距[3],这些问题推动了计算方法的发展.当前许多方法考虑联合蛋白质多种特征共同对蛋白质的功能进行预测,然而对于未知的蛋白质来说除序列外其他特征难以获得,目前许多方法忽略了蛋白质标签之间存在的联系,本文在对比学习的框架下进一步挖掘蛋白质序列信息,同时也考虑到了蛋白质标签之间的联系.
1 相关工作
蛋白质功能预测是生物信息学中的一项重要任务,对于揭示蛋白质在病理生物学中的作用和寻找药物靶点具有重要意义.随着高通量测序技术的发展,快速便捷地获取蛋白质序列的同时,也发掘出大量未知功能的蛋白质序列,早期基于定量生化实验验证蛋白质功能的方法已经无法满足目前的需求,为了高效而准确地预测蛋白质功能,不同的研究者提出了不同的方法,这些方法可归纳为基于信息的方法和基于机器学习的方法2个方向.
1.1 基于信息的方法
同源蛋白质的序列通常是高度相似的,因此可能具有非常相似的功能.通过将新发现的蛋白质序列与已经被注释的蛋白质序列进行比对,选取与新发现的蛋白质序列相似性最高的已知蛋白质序列,从而推断出新发现的蛋白质所具有的功能.序列比对的正是基于同源序列具有相似功能的原则对未知蛋白质功能进行预测.双序列比对是序列比对中常用的一种方式,它的匹配方式分为2种:一种是局部序列比对,常用工的具为Blast,Blast通过将一条序列分割为多个短片段,并在数据库中搜索与这些片段匹配的序列,从而寻找最佳匹配的序列[4];另一种是全局比对算法Needleman-Wunsch,该算法通过对2条序列进行动态规划来寻找最佳比对,并根据一定的打分规则对所有可能的比对方式进行评估,最终在两条序列间找到最佳匹配[5].多序列比对是序列比对中常用的另一种方式,在多序列比对过程中,若3条或3条以上的序列具有共同的祖先,则通过比较这些序列的相似性,可以推断出它们在进化树上的位置,进而推断它们是否具有同源关系,常用的多序列比对工具为Muscle[6].
结构域是构成蛋白质的基本结构单元之一,具有独立的结构和功能.不同的结构域可以执行特定的功能从而影响蛋白质的功能,通过识别蛋白质序列中包含的结构域可以预测蛋白质的功能.Mistry等[7]基于隐马尔可夫模型识别出未知蛋白质序列中所包含的结构域,并将其与蛋白质家族数据库中的结构域进行比对,从而预测出未知蛋白质的功能.
蛋白质的功能不仅仅取决于氨基酸序列,往往也与三维空间结构密切相关.例如胰岛素和胰高血糖素这2种激素,尽管它们的氨基酸序列存在较大差异,但是它们的结构非常相似,这种相似性导致它们具有相似的功能[8].由此可以通过预测蛋白质的结构进而预测蛋白质的功能,蛋白质结构预测方法通常被归类为基于模板的建模和无模板建模方法,基于模板的建模方法通过复制和提炼现有蛋白质的结构框架来构建模型从而预测蛋白质结构,而无模板建模的方法则不使用全局模板结构来预测蛋白质结构[9].此外还有一些其他方法,例如李绍新等[10]采用遗传算法来预测蛋白质结构.
基于信息的方法利用蛋白质序列比对、蛋白质序列性质以及蛋白质结构等信息来推断蛋白质的功能,从蛋白质信息对蛋白质的功能进行预测的方法是通用的,然而数据库中需要存储有与未知蛋白质功能相似的已知蛋白质.
1.2 基于机器学习的方法
多年前研究人员已经开始利用机器学习技术来对未知蛋白质进行功能预测并显示出良好的性能.Maheshwari等[11]训练了一个朴素贝叶斯分类器来预测蛋白质与蛋白质之间的相互作用,该方法使用了蛋白质序列和结构信息作为输入特征,并使用朴素贝叶斯分类器对蛋白质功能进行预测,然而该方法对输入数据的质量要求较为严格.Fabris等[12]使用了扩展的局部层次朴素贝叶斯算法对蛋白质功能进行预测,然而该算法复杂度较高、可解释性较差.Makrodimitris等[13]使用了KNN算法对蛋白质功能进行预测,K最近邻算法是一种非参数分类方法,通过对给定特征空间中最近的k个点的标签进行多数投票从而对给定观测点进行预测分类,然而当训练数据的标签分布不平衡或存在偏差时,多数投票的结果将会受到影响.
Mansoor等[14]采用对抗神经网络结构来对蛋白质进行功能预测,该方法在模型预训练阶段训练一个生成器和判别器,预训练结束之后丢弃生成器,保留判别器并利用判别器对蛋白质进行功能预测.Elhaj等[15]提出了一种混合深度神经网络模型,该模型对卷积神经网络和长短期记忆神经网络进行集成操作后对蛋白质序列的特征进行提取,最终对蛋白质功能进行预测.Borges等[16]使用竞争神经网络进行多标签分层分类,与标准多层感知器的区别在于输出层的神经元使用竞争性激活函数激活,因此只有一个输出神经元将被成功激活,而其他神经元则被抑制.概率神经网络使用贝叶斯最优决策规则进行分类,并考虑每个类别的概率密度函数,模型SVM-Prot使用了概率神经网络,在从序列中识别蛋白质功能方面表现出了比KNN和SVM更好的预测性能[17].Zacharaki等[18]使用CNN来结合SVM以及KNN分类器来预测蛋白质功能,该方法显著优于单一使用KNN的方法对蛋白质进行功能预测.ProTranslator模型从蛋白质序列信息、文本信息以及结构信息多个角度挖掘了蛋白质特征,有效提高了蛋白质功能预测模型的准确性[19].
模型DeepGO使用卷积神经网络和循环神经网络结合的方式,从蛋白质序列和蛋白质进化信息中提取特征,进而对蛋白质的功能进行预测[20].模型deepNF使用深度网络融合的方法对蛋白质进行功能预测,该模型使用多模式深度自动编码器提取特征,然后将其传递给SVM[21].模型DeepGOZero使用了一种全新的模型结构来预测蛋白质功能,其模型包含一个用于蛋白质表示学习的自编码器网络以及另一个用于多标签分类的多层感知器网络[22].递归神经网络适合于处理顺序数据,长短期记忆网络是递归神经网络的一种进化,模型Deepcnnlstmgo将卷积神经网络和长短期记忆神经网络进行集成,从氨基酸序列中学习特征后对蛋白质功能进行预测,取得了不错的预测效果[23].王皓白等[24]提出的蛋白质自编码器模型,称为Protein-HVGAE,该模型使用2个双曲图卷积网络作为编码器,通过在不同曲率的双曲空间中计算隐藏层的均值和方差,从而捕捉网络的层次结构,这种方法有助于区分各节点的低维表示,更好地描述蛋白质的特征.
蛋白质功能预测本质上是多标签分类问题,输入需要映射到离散输出.基于机器学习的方法关注学习数据独特性,这样模型可以自动发现数据的重要特征,由此对数据内容进行功能预测.因此基于机器学习的方法可以更好地预测未知蛋白质的功能,然而目前绝大多数机器学习方法没有充分挖掘蛋白质序列信息的同时关注蛋白质标签之间存在的联系.
2 方法
本文提出一个基于对比学习框架的蛋白质功能预测模型用于预测蛋白质具有的功能,总体框架如图1所示.整个过程分为4个阶段:数据预处理阶段、特征提取阶段、模型预测阶段以及标签传播阶段.其中,数据预处理阶段、特征提取阶段以及模型预测阶段用于模型训练,在预测阶段得到初步预测结果,标签传播阶段则是在初步预测结果的基础上进行的,旨在进一步提高模型预测的准确性.
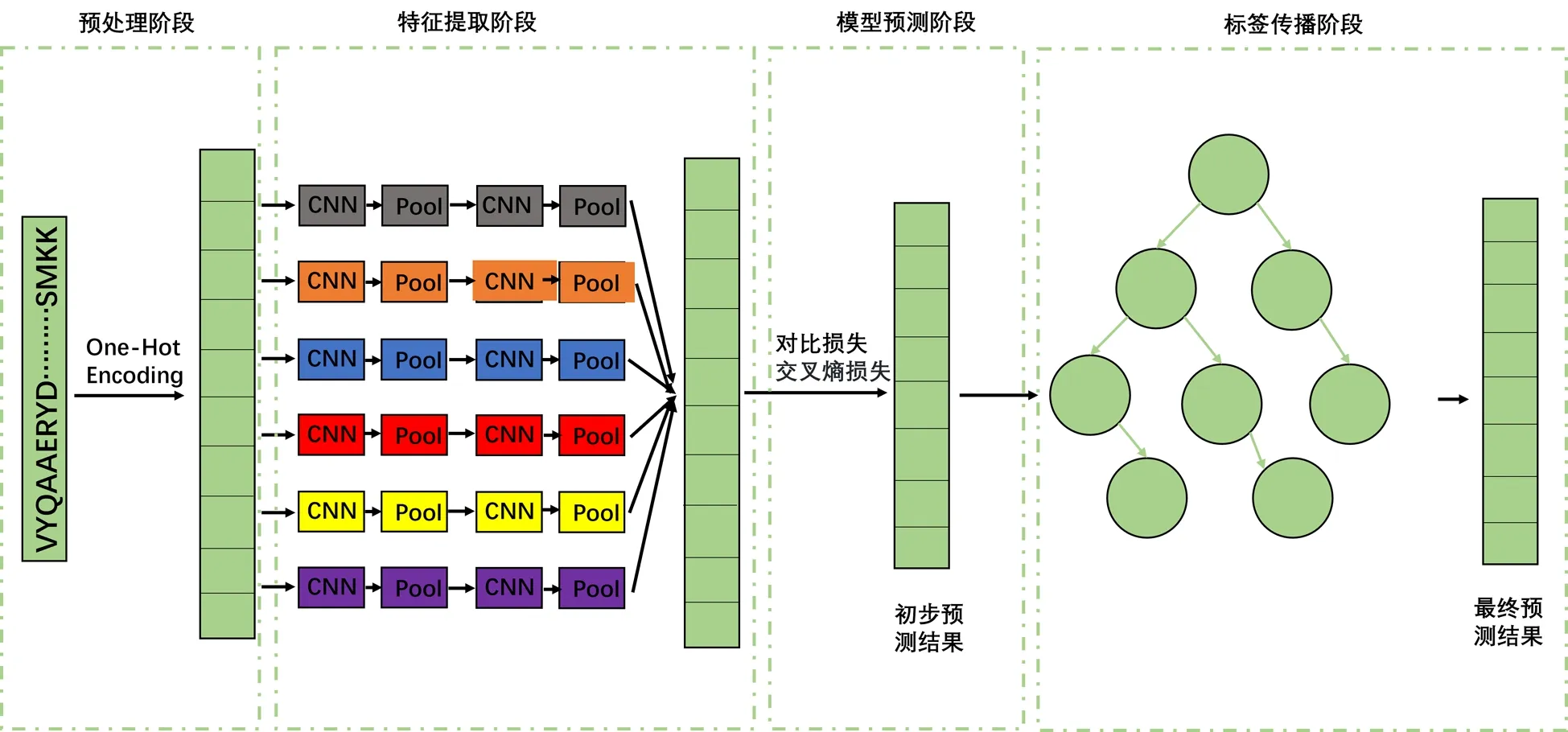
图1 基于对比学习的蛋白质功能预测模型Fig.1 A protein function prediction model based on contrastive learning
2.1 数据预处理阶段
蛋白质序列由20个氨基酸组成,这些氨基酸组成了现实生活场景中的多数蛋白质序列,由于计算机不能直接处理蛋白质的氨基酸序列,因此需要对每条蛋白质序列进行编码.
本文将每个氨基酸视为一个独热码,并使用独热码的组合来表示蛋白质序列,模型的输入是由氨基酸的不同独热码所组合.每个氨基酸都可以通过一个长度为21的独热码向量来表示,其中一个独热码用来表示除20种氨基酸之外出现的其他异常值.每条蛋白质序列长度各不相同,因此对超过1 000个氨基酸长度的蛋白质序列取前1 000个氨基酸,对小于1 000个氨基酸长度的蛋白质序列补零至1 000个氨基酸长度,最终得到了大小为21×1 000的特征向量,每条蛋白质序列可以用以下矩阵表示:
Xi=[xi1,xi2,…,xi1000],
其中,Xi∈21×1 000,i代表数据集中的第i个蛋白质,xij代表第i个蛋白质的第j个氨基酸的独热编码.
2.2 特征提取阶段
在神经网络模型中,本文定义了一个长度为m的卷积核列表K,对每个大小为k∈K的卷积核,定义一个卷积神经网络层f,见式(1):
(1)
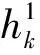
通过式(1),得到m个不同的特征向量,将所有的特征向量拼接在一起,得到向量Y:
Y=f1(X)⊕f2(X)⊕f3(X)…fm(X),
其中,fm(X)表示第m个卷积神经网络层,X表示输入数据,⊕表示向量拼接.
2.3 模型预测阶段
模型预测阶段是基于特征提取阶段获得的特征向量对蛋白质序列进行功能预测的过程.预测阶段应用在训练任务和预测任务中,在训练任务中探讨如何将2种损失函数结合在一起进行模型优化,在预测任务中则是使用训练后的模型对评估数据集进行评估进而得到初步预测结果.
在特征提取阶段,本文获得序列特征向量Y,之后通过一个全连接层将特征向量映射到输出类别空间并进行激活,得到预测的标签向量Z,具体过程见式(2):
Z=σ(MLP(Y)),
(2)
其中,σ(·)表示sigmoid函数,MLP表示线性变换,Y表示输入向量,最终输出的标签向量Z中的每个元素代表对应类别的预测概率.
在训练任务中,损失函数是用来衡量模型预测输出与真实值之间差异的一种函数,通过最小化损失函数来对模型进行训练和优化.对比损失和交叉熵损失是深度学习中常用的2种损失函数.交叉熵损失函数主要用于分类问题,衡量模型对于样本分类的准确程度,而对比损失函数则主要用于度量样本之间的相似度和差异度.2种损失函数结合使用可以更有效地提高模型的分类精度、鲁棒性和泛化能力.
交叉熵是用于衡量模型的预测的输出与真实值之间的差异.在蛋白质功能预测任务中,使用二元交叉熵作为损失函数,计算所有蛋白质功能预测的交叉熵损失平均值,即:
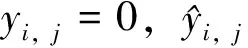
监督对比学习的目的是通过缩小同一类实例之间的距离,同时增大不同类实例之间的距离来实现分类任务.在蛋白质功能预测任务中不同的蛋白质序列可能会共享一些注释,同时每条蛋白质序列也可能具有一些特有的注释.因此在应用对比学习的方法对蛋白质进行功能预测时,如何利用这类特性是关键问题.本文提出了一种基于标签相似度的方法,根据蛋白质序列的标签相似度动态调整实例之间的距离权重,更好地处理共享标签和特有标签之间的关系.
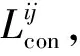
其中,d(·,·) 表述余弦相似度,通过计算两个向量之间的夹角余弦值所得,τ表示对比损失函数的温度系数,设置为1.xi表示蛋白质序列表示向量,它是通过式(2)计算所得.αij是一个正则化系数,用于调整不同样本之间的对比损失重要性,计算方式如下:
其中,Si,j表示蛋白质序列i和蛋白质序列j标签向量之间的相似度,αij表示标签相似度的正则化.
最后,所有序列对的平均对比损失表达如下:
其中,n表示蛋白质序列的数量.
将两个损失相加得到整个模型训练阶段的总损失:
L=λLBCE+ (1-λ)Lcon,
其中,λ是一个超参数,本文设置为0.7.
在预测任务中,损失函数通过验证数据集计算真实值与模型预测输出概率之间的差距来评估模型当前预测能力的好坏,并通过优化算法来更新模型参数以使模型性能得到提升.最后通过使用训练优化好的模型对评估数据集中的每条蛋白质序列进行评估,得到初步预测结果.
2.4 标签传播阶段
Gene Ontology(GO)是一种广泛使用的蛋白质功能注释系统,它提供了一套标准化的术语和分类体系,GO术语是一组标准化的词汇,用于描述蛋白质在分子功能、生物过程和细胞组成这3个不同层面表达的功能,一条蛋白质序列可以由多个GO术语描述,即多个标签,并且多个GO术语被组织到一个有向无环图中,因此本文利用多个GO术语之间的结构关系,对初步预测得到概率较高的GO术语进行传播,以提高预测概率较低的GO术语,进而提高模型的预测准确率.
为了提高蛋白质序列预测的GO术语f的可靠性,需要考虑该术语在有向无环图中祖先节点的预测结果,定义Af为术语f在有向无环图中的所有祖先节点的预测术语(包括f自身)的集合,Af中每个术语的预测为pa.Af中所有术语的平均预测为pf,即:
其中,|Af| 表示集合Af的元素个数.
在蛋白质功能预测的任务中,集合Af中的所有GO术语共现存在,即要么同时作为一条蛋白质序列标签出现,要么同时不出现,在Af中存在2种概率,一种是自信的预测概率,即高于pf的pa,另一种是不自信的预测概率,即低于pf的pa,本文考虑如何利用自信预测概率影响不自信预测概率来提高蛋白质功能预测模型的准确性.
定义一个自信区间(pf,2pf]来判断哪些节点为自信预测概率节点,并给自信预测概率定义一个上限阈值2pf,目的是为了防止噪声预测概率对Af中其他节点的预测概率产生的消极影响.通过传播自信预测概率来影响比自信预测概率低的pa,影响方式为将低于自信预测概率的pa替换成自信预测概率的pa,同时在这个过程中,不会影响原来就比自信预测概率高的pa.整个传播过程的结果以字典键值对的关系进行存储,当对一条蛋白质序列的所有预测概率传播结束之后,用传播后的所有预测概率替换原来的预测概率,从叶子节点向根部节点的方向进行预测概率的传播,这样保证了在传播过程中对Af中更多数量的pa产生积极影响.
3 实验及分析
3.1 数据集
本文使用2016年9月发布的CAFA3挑战的训练数据集,以及使用2017年11月15日发布的评估数据集来评估本文的蛋白质功能预测模型[25].训练集包含截至2016年9月之前已知的所有具有实验标注的蛋白质,测试集则包含在2016年9月至2017年11月之间获得实验标注的蛋白质,其具体内容见表1.

表1 本体类型及相应的数据集大小Tab.1 The types of ontologies and their corresponding dataset sizes
基因本体的层次结构在生物信息学的研究中被广泛应用,并帮助研究人员进行蛋白质功能注释[26].本文使用的基因本体发布于2016年6月,拥有11 173个Molecular Function(MFO)类、29 542个Biological Process(BPO)类和4 082个Cellular Component(CCO)类,该版本被用于评估CAFA3预测.
3.2 实验参数说明
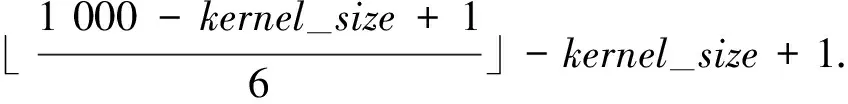
全连接层的输入特征向量的大小,这里是1 024×6,由每个卷积层的输出特征展平成一个一维向量拼接起来所得,最后的输出向量通过sigmoid激活函数激活.
3.3 评价指标
在这项研究中,本文使用Fmax、AvgPr和AvgRc来评估模型的性能。Fmax是一个以蛋白质为中心的最大F值,AvgPr是至少预测出一个GO术语的所有蛋白质序列的平均精度,AvgRc是至少具有一个真实GO术语的所有蛋白质的平均召回率,计算公式如下:
其中,当阈值为t时,Pi(t)表示蛋白质序列i在阈值为t的条件下预测出的GO术语的集合,Ti表示蛋白质序列i真实的GO术语集合,n是参与评价的所有蛋白质的数量,I是一个判别函数,若符合I规定的判定条件则返回1,否则返回0.
3.4 实验及分析
在基准评估数据集上将本文的模型与之前的CAFA3挑战中表现优异的DeepGO[20]、FFPred3[27]、DeepGOA[28]和Phylo-PFPtextsuperscript[29]模型进行了比较,所有方法在训练过程中都没有使用评估数据集中的蛋白质注释,比较结果如表2所示.
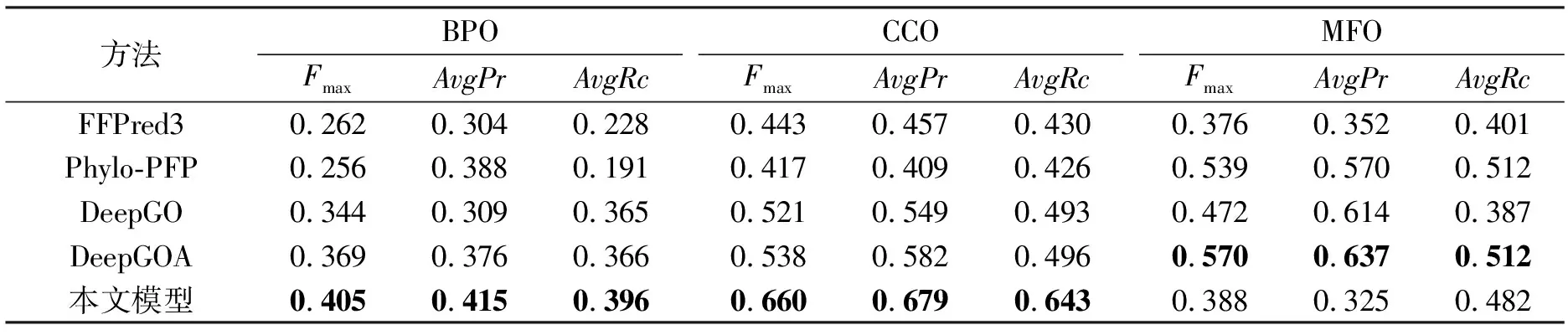
表2 不同方法在3个生物过程上的表现比较Tab.2 Performance comparison of different methods on three biological processes
观察表2的结果,本模型在预测蛋白质的生物过程(BPO)方面,所有指标上都优于其他模型,这表明本文模型具有较强的生物过程预测能力,在预测蛋白质的细胞组成(CCO)方面,本文模型的评价指标也优于其他模型.然而在预测蛋白质的分子功能(MFO)方面,评价指标相对于其他模型表现较差,其主要原因是在3个生物过程中,蛋白质的分子功能的训练数据最少,导致模型学习到的特征表示不够充分,进而难以捕捉到多个功能之间的复杂关系,最终表现在模型预测性能下降.
综上所述,本模型的主要优势是能更精准地预测蛋白质的生物过程和细胞组成,在分子功能预测能力方面还需要进一步改进.尽管本文提出的模型在蛋白质功能预测任务上已经取得了很大进步,但在分子功能预测方面仍需要进一步优化,以提高整体的预测效果.
3.5 总结与展望
本文提出了一种基于对比学习的蛋白质功能预测模型.该模型首先使用大小不同的卷积核对蛋白质序列进行提取特征,之后在对比学习框架下进一步提取蛋白质序列特征,最后基于初步预测结果,利用GO术语之间存在的共现关系,对预测结果进行传播和增强,进而得到最终的预测结果并且进一步提高了蛋白质功能预测模型的准确性.
未来将解决蛋白质功能预测过程存在的过拟合问题.蛋白质功能预测实际上是一个多标签分类任务,其训练样本数量无法涵盖所有标签组合空间,因此下一步将设计合适的模型来提高蛋白质功能预测模型的泛化能力,进一步提高预测准确性.
本文代码地址:https:∥github.com/sxdzh666/A-protein-function-prediction-model-based-on-contrastive-learning.
猜你喜欢
中学生数理化·中考版(2022年6期)2022-06-05 06:49:10
中学生数理化·中考版(2021年6期)2021-11-22 07:52:30
新世纪智能(数学备考)(2021年4期)2021-08-06 09:04:50
新世纪智能(数学备考)(2021年4期)2021-08-06 09:04:50
车迷(2018年11期)2018-08-30 03:20:32
海峡姐妹(2018年3期)2018-05-09 08:21:02
公民与法治(2016年10期)2016-05-17 04:12:58
计算机工程(2015年8期)2015-07-03 12:20:27
中国科技术语(2012年3期)2012-03-20 14:36:13
中国科技术语(2012年3期)2012-03-20 14:36:11
