
Therapeutic potential of iron chelators in retinal vascular diseases
2023-11-10 04:05:14YanLiZiXuanChengTingLuoHongBinLyu
Yan Li, Zi-Xuan Cheng, Ting Luo, Hong-Bin Lyu
1Department of Ophthalmology, the Affiliated Hospital of Southwest Medical University, Luzhou 646000, Sichuan Province, China
2Department of Ophthalmology, the People’s Hospital of Jianyang, Chengdu 641400, Sichuan Province, China
Abstract
● KEYWORDS: iron overload; oxidative stress; inflammation;blood–retinal barrier; ferroptosis; iron-chelating agent
INTRODUCTION
Iron is the basic component of hemoglobin and ferrithin protein, which is responsible for oxygen transport, cellular metabolism, and deoxyribonucleic acid synthesis in the human system.Iron can complete electron transfer while transiting between ferrous (Fe2+) and ferric (Fe3+) states, enabling it to be widely involved in various metabolic pathways[1], such as the production, differentiation, and proliferation of cellular energy,and as a helper cofactor of mass enzymes[2].Iron is crucial for the retina since it is one of the body’s most metabolically active tissues.Trace amounts of iron are essential for cell survival.However, excessive iron will promote a large amount of reactive oxygen species (ROS) through the Fenton reaction,which can augment the activation of the hypoxia-inducible factor 1α (HIF-1α).ROS includes superoxide anions (O2-),hydrogen peroxide (H2O2), superoxide radicals (ROO·), and hydroxyl radicals (-OH), which are commonly considered toxic to cells[3].The other major aspect of iron toxicity is nuclear factor kappa B (NF-κB) activation[4].NF-κB promotes the production of cytokines, adhesion molecules, and growth factors, ultimately triggering inflammation.The transcription factor NF-κB is recognized as a crucial transcription factor that is effectively activated by HIF-1α[3].HIF-1α induces genes,such as vascular endothelial growth factor (VEGF), which is needed to maintain retinal homeostasis under hypoxia.VEGF is widely acknowledged as a significant component that triggers pathological neoangiogenesis in the retina[5].The accumulation of free Fe2+in the cell membrane can lead to ferroptosis, a type of iron-dependent regulatory cell death named in 2012, characterized by excessive accumulation of iron, excessive peroxidation of phospholipids containing polyunsaturated fatty acids (PUFAs), increased ROS and decreased glutathione (GSH) and glutathione peroxidase 4(GPX4) levels[6-7].Iron chelators [e.g., desferriamine (DFE),deferasirox (DFX),etc.] as well as lipid ROS scavengers (e.g.,ferrostatin 1) have been shown to possess inhibitory effects on this form of cell death[8-11].Currently, there is evidence of iron overload and ferroptosis in several retinal vascular illnesses[12-15].It is suggested that ferroptosis might have a significant impact on inflammation and damage to retinal vascular endothelial cells generated by oxidative stress[12].Iron chelators have the ability to hinder the Fe2+-induced Fenton reaction, hence mitigating oxidative stress[16].Furthermore, the anti-inflammatory properties of iron chelation safeguard the retina, even when iron toxicity is not the primary factor[17].So,using iron chelators to improve problems with retinal vascular endothelial dysfunction has a lot of potential[18-19].
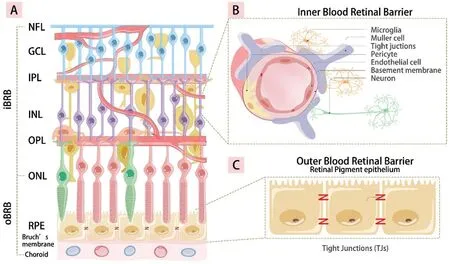
Figure 1 The iBRB and oBRB in the retina A: The structure of the retina; B: The iBRB is composed of the tight connection between retinal capillary endothelial cells, pericytes, astrocytes, Müller glial cell, and microglials; C: The monolayer tight connection between RPE cells and villous capillaries constitutes of the oBRB.iBRB: Inner blood-retinal barriers; oBRB: Outer blood-retinal barriers; NFL: Nerve fiber layer; GCL:Ganglion cell layer; IPL: Inner plexiform layer; INL: Inner nuclear layer; OPL: Outer plexiform layer; ONL: Outer nuclear layer; RPE: Retinal pigment epithelium.
IRON IN RETINA
Absorption and Transport of Iron in RetinaTwo bloodretinal barriers (BRB) separate the retina from blood circulation.The inner BRB is composed of the tight connection between retinal capillary endothelial cells, pericytes,astrocytes, Müller glial cells, and microglials[20].Retinal pigment epithelium (RPE) cells are in close contact with villus capillaries, and the tight monolayer connection between them constitutes the outer BRB (Figure 1)[12,21].Additionally, both the inner and outer BRBs serve to prevent excessive iron influx into the eye in instances of cyclic iron overload[12].Through[transferrin (TF) receptor 1] (TFR1) on the RPE basement membrane, chorionic capillaries send iron that is bound to TF to the RPE.The outer segment of photoreceptors (PRs)contains a large amount of iron.Another way for RPE to ingest iron is to phagocytize the outer segment of the PRs[22].After entering the RPE cytoplasm, iron is stored in melanosomes and ferritin[23].In the inner layer of the retina, TFR1 is expressed on the lumen side of retinal endothelial cells, and iron is transported to the cell through TFR1[24].Iron output depends on the release of iron and ferroportin (FPN) from the core[25].There are two distinct levels at which iron homeostasis occurs:cellular and systemic[26].The iron regulatory proteins (IRPs),specifically IRP1 and IRP2, have a significant function in cellular iron homeostasis through their regulation of key ironrelated proteins, including TFR-1, divalent metal transporter-1(DMT1), FPN1, and ferritin.When cellular iron levels are depleted, IRP1 and IRP2 identify and attach to iron responsive elements (IREs) located in the untranslated regions (UTRs)of messenger RNAs (mRNAs), which encode the translation of various proteins involved in regulating iron metabolism[27].In contrast, it has been observed that IRP is unable to form a complex with IRE42 located in the UTR of these mRNA molecules under conditions of elevated cellular iron leves[28].The local homeostasis of retinal iron is independent of systemic regulation due to the separation of the BRB.
Physiological Role of Iron in RetinaIron in the retina is mainly located in the RPE, PR, and choroid[29].Iron is one of the important metals in the retina[30], involved in the energy metabolism of retinal cells[31]and the secretion of neurotransmitters[32].Iron is a key component of many retinal enzymes, which are essential for maintaining retinal function[33].It is an important cofactor of the RPE-specific 65 kDa protein, which is involved in phototransduction and visual adaptation by regulating the metabolism of retinoids to modulate chromophore levels in light-harvesting PRs[34].Fe2+, as a cofactor of prolyl hydroxylase, participates in the expression and degradation of HIF[18,35].Conversely, HIF acts as a transcription factor for some iron homeostasis genes(e.g., TF, TFR1,DMT1, FPN, and ceruloplasmin genes) to participate in iron metabolism by binding to specific hypoxia response element (HRE) sites on mRNA[12].HIF induces VEGF to participate in the physiological and pathological angiogenesis of retina[5,36].Health and pathological retinal status exhibit a strong correlation with iron levels[2,22].
Iron Overload and Retinal Vascular InjuryOxidative stress, inflammation, ferroptosis, and angiogenesis may be the main causes of the increased retinal iron and the damage to the BRB that follows.
Iron overload and retinal oxidative stressElevated levels of iron within the eye have been observed to contribute to the occurrence of oxidative stress and subsequent inflammatory damage.The retina is a lipid-dense tissue containing high concentrations of PUFAs[37], making it particularly vulnerable to ROS-induced oxidative damage[13].On the one hand,hypoxia leads to increased iron uptake through the increased expression of cell iron input genesDMT1andTFR1mediated by HIF[38].Oxidative stress can also upregulateTFR1[39].The inflammatory factor interleukin (IL)-6 has been proven to mediate the upregulation of hepcidin to induce cell iron isolation[40].Conversely, cellular conditions characterized by low pH and elevated concentrations of superoxide and peroxides can induce the liberation of iron from its storage protein.Consequently, this process results in an augmentation of the labile iron pool (LIP), primarily consisting of redoxactive Fe2+, which will intensify the production of harmful ROS[41].In a particular investigation, Rogerset al[42]administered intravitreal injections of iron into the ocular region of adult C57BL/6 mice.The results revealed an elevation in retinal ROS levels, disruption of the outer retinal nuclear layer nucleus, rapid PR cell death, and the subsequent development of both map atrophy and sympathetic ophthalmia[43].Hope-Rosset al[44]studied eight patients with iron foreign bodies retained in the eyes after trauma.The presence of iron can lead to the generation of free radicals, resulting in the occurrence of oxidative stress.All of the patients have varied degrees of impaired vision.Due to proliferative vitreoretinopathy, the last vestiges of eyesight in two individuals were limited to light perception.Research has shown that retinal endothelial cells are more susceptible to oxidative damage compared to endothelial cells in other locations[45].
Iron and retinal inflammationVarious inflammatory and immunological diseases are caused by problems in iron metabolism, which can raise the quantity of iron inside cells[46].The NF-κB signal pathway can be activated by excess iron[47-48],which encourages the creation and release of inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and IL-1β[49].TNF-α injections into the eye can significantly damage the BRB and raise endothelial cell permeability[50].Due to research, IL-1β has an aspect in how the inner BRB breaks down[51].Elevated levels of IL-6 promote an upregulation in the synthesis of hepcidin, resulting in the destruction of iron transport proteins and subsequently causing an augmentation in intracellular iron levels[46].The NRLP3 inflammasome pathway is activated in RPE cell due to an abundance of intracellular iron.This activation occurs as a result of the inhibition of aluRNA degradation by double-stranded RNA-specific endoribonuclease.There may be a reciprocal connection between iron metabolism problems and inflammation.
Iron overload and retinal programmed cell deathApoptosis was produced experimentally by injecting iron particles into the vitreous cavity of rats, as evidenced by the presence of TUNEL-positive nuclei, particularly in the outer nuclear layer,after 48h.In a mouse model of retinal ischemia reperfusion(IR), TF dramatically rose 2h after ischemia, peaked at 12h,and then progressively dropped, indicating that intracellular iron excess occurred at an early stage.Retinal ganglion cells(RGCs) initially experience necrosis during the process of retinal IR damage, followed by apoptosis after some time, and subsequently ferroptosis completes the process[52].Blocking the ferroptosis genes can significantly reduce the damage and enhance the survival of RGCs.Apoptosis inhibitors(z-VAD-FMK), necrosis inhibitors (necrostatin-1), and ferroptosis inhibitors (ferristatin-1) were used to interfere with experimental mice and primary cultured RGCs, respectively.They showed a significant anti-IR protective effect of RGCs,among which ferristatin-1 had the best therapeutic effect,suggesting that ferroptosis plays a more important role in the death process of RGCs[53].
Iron overload and retinal vascular angiogenesisTheFe2+ion serves as a cofactor for proline hydroxylase, contributing to the regulation of HIF expression and degradation and so exerting a significant influence on the process of angiogenesis[18].The elevation in iron concentrations in the retina might potentially facilitate the upregulation of succinate receptor 1 through the suppression of the anti-angiogenic properties of cleared high-molecular-weigh kininogen (Hka).Consequently, this mechanism may induce the synthesis of angiogenic molecules.The involvement of ROS-induced elevation of VEGF level is crucial in the pathogenesis of ocular diseases, leading to increased vascular permeability and disruption of the BRB[54].Excessive ROS leads to the elevation of HIF-1αviaoxidative stress.Consequently, the enhanced transcriptional control exerted by HIF-1α results in the upregulation of VEGF expression[55].The excessive generation of ROS and the overexpression might result in the impairment of both paracellular and transcellular endothelial transport mechanisms[56].VEGF exerts its inhibitory effect on tight junctions by binding and activating two specific tyrosine kinase receptors, namely VEGFR1 (Flt-1) and VEGFR2(KDR/FLk-1).This activation then leads to a reduction in the expression levels of relevant proteins.VEGF assumes a significant role in the pathogenesis of BRB dysfunction[57]as well as in the regulation of pathological angiogenesis[58].VEGF and inflammatory factors have been shown to upregulate the expression of vascular cell adhesion molecule-1 (VCAM-1)and intercellular adhesion molecule-1 (ICAM-1), therefore promoting the adherence of leukocytes to the vascular wall[47-51].Furthermore, the process of rearrangement and migration of endothelial connexins, including vascular endothelial cadherin and tight junction proteins, into tissues is facilitated by VCAM-1 and ICAM-1[59].This phenomenon ultimately results in endothelial cell damage and the destruction of BRB[60].Retinal edema and visual loss occur after increased retinal vascular permeability[61].Retinal iron excess has been linked to direct BRB injury in studies (Figure 2)[62].Compared with nondiabetic mice, diabetic mice have increased iron content in the retina, loss of retinal barrier integrity, and abnormal vascular changes such as vascular curvature, arteriovenous crossing,and retinal vein occlusion[62].Mice lacking ceruloplasmin have subretinal neovascularization, retinal degeneration, and retinal iron overload[63].In a recent study on oxygen-induced retinopathy[52-53,64], it was established that ferroptosis has a role in the process of pathological angiogenesis[52-53,64].Therefore,the targeted inhibition of ferroptosis can effectively protect the retina[65].
IRON OVERLOAD IN RETINAL VASCULAR DISEASE Diabetic RetinopathyDiabetic retinopathy (DR) is the main cause of vision loss worldwide and represents a significant consequence associated with diabetes[66].Clinically, DR can be divided into non-proliferative DR (NPDR) and proliferative DR (PDR) according to whether retinal neovascularization occurs.In diabetes, long-term hyperglycemia leads to the accumulation of many metabolic substances in cells, such as advanced glycation end-products (AGEs) and protein kinase C, as well as the renin-angiotensin system, and the release of a large number of inflammatory factors, including IL-1β, IL-6, IL-8, and TNF-α.The increased inflammatory factors can damage the retina by promoting angiogenesis and neurodegeneration, accompanied capillary occlusion,vascular leakage, retinal ischemia and injury, and hypoxia[67].Studies have shown that oxidative stress increases HIF-1α by producing a large amount of ROS in hyperglycemia to release VEGF[68].Increased retinal VEGF expression causes BRB damage, leading to increased vascular permeability,ischemia-induced formation of new blood vessels, ultimately,PDR[69].It is reported that the retinal iron content is increased in postmortem retinal samples collected from patients with diabetes and diabetic animal models[62].Clinical samples also suggest a close relationship between iron contentin vitroand PDR[70].Previous studies have proved that vascular endothelial cells are sensitive targets of hyperglycemia[71].Intriguingly,the process of ferroptosis and the endothelial dysfunction induced by diabetes have many similar characteristics, such as the accumulation of ROS and the enhancement of oxidative stress[72].Under hyperglycemia, the total ROS and intercellular ROS in human retinal vascular endothelial cells (HRECs) are increased, and the expression of GPX4 is significantly reduced,suggesting that endothelial cells have oxidative stress under high glucose and that ROS-induced lipid peroxidation initiates ferroptosis[73].HRECs treated with high glucose promote GPX4 ubiquitination by increasing tripartite motif-containing protein 46 (TRIM46) expression, contributing to the ferroptosis of HRECs induced by high glucose[74].This programmed cell death can be reversed by a ferroptotic inhibitor.The ferroptosis inhibitor liproxstantin-1 (LX-1) has a strong protective effect on retinal function in diabetes rats[75].The observation of strong iron markers in RPE and the outer reticular layer in patients with DR is strong evidence of the iron-related death of PRE[76].High glucose increases intracellular Fe2+concentrations to induce ferroptosis in ARPE-19[77], as indicated by decreased glutathione, increased malondialdehyde, and decreased cell survival[78].At present, ferroptosis has been studied as an early treatment target for DR[15].DR is a metabolically related disease regulated by multiple genes in which multiple metabolic processes, including iron metabolism, glucose metabolism, and lipid metabolism, are also involved.The molecules and pathways related to ferroptosis are complex and require further research.
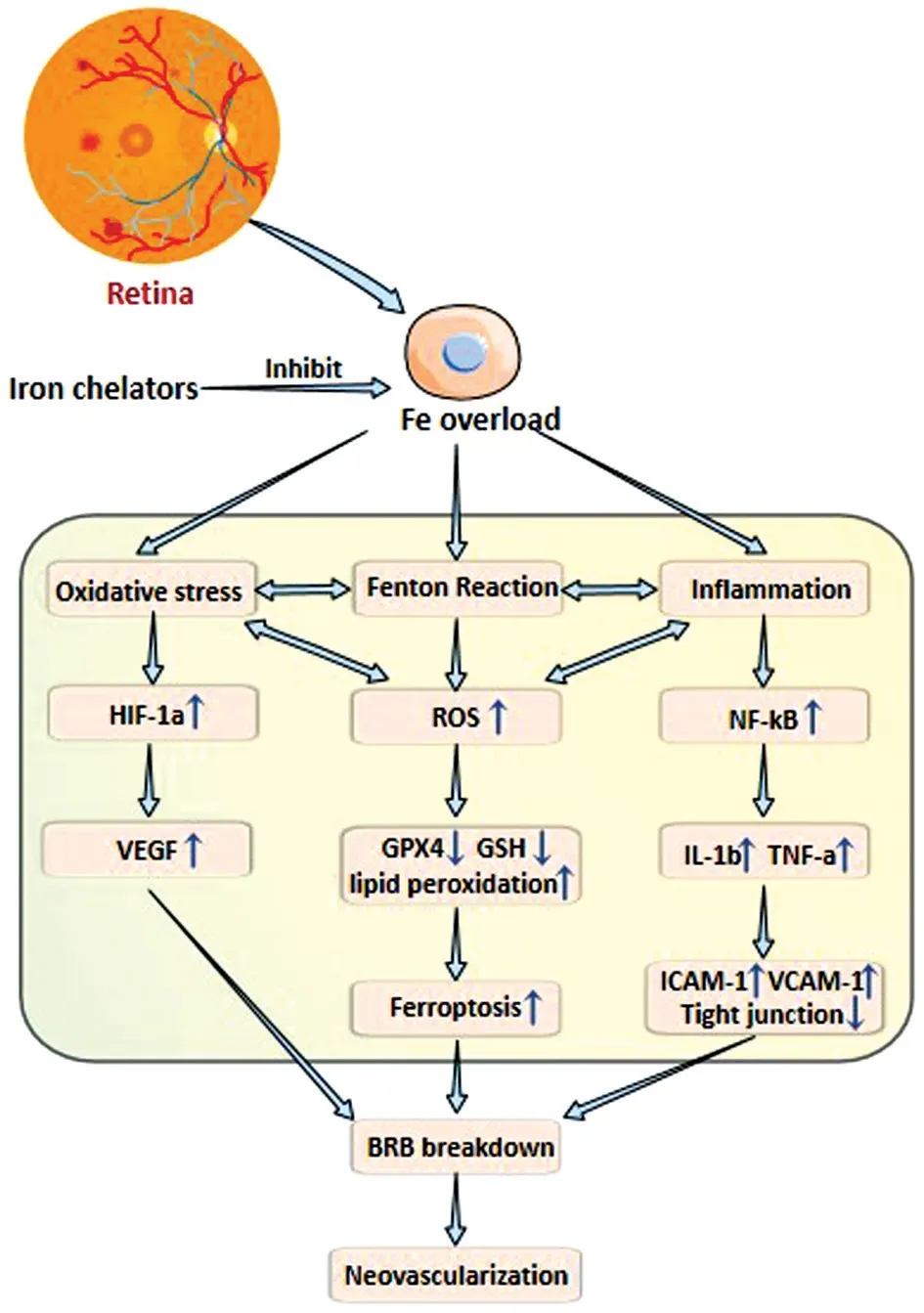
Figure 2 Iron chelators reduce oxidative stress and inflammation caused by iron overload, protect BRB and inhibit pathological angiogenesis HIF-1α: Hypoxia-inducible factor 1α; ROS: Reactive oxygen species; NF-κB: Nuclear factor kappa B; VEGF: Vascular endothelial-derived growth factor; GPX4: Glutathione peroxidase 4;GSH: Glutathione; IL-1: Interleukin-1; TNF-α: Tumor necrosis factoralpha; VCAM-1: Cell adhesion molecule-1; ICAM-1: Intercellular adhesion molecule-1; BRB: Blood-retinal barriers.
Retinal Vascular OcclusionsRetinal vein occlusion (RVO)is the second most prevalent cause of visual deterioration among retinal vascular diseases, ranking only after DR[79].In a study comparing the retinal blood flow velocity of healthy individuals and branch retinal vein occlusion (BRVO) or central retinal vein occlusion (CRVO) patients, the researchers found that the lower the blood flow velocity, the more serious the RVO[80].Decreased blood flow may induce inflammation,leukocyte adhesion, and retinal endothelial barrier damage through the activation of the Krüppel-like factor of B cells,NF-κB, and other mediators[51].In 1995, Itoet al[81]incubated methemoglobin with retinal homogenatein vitro.Atomic absorption analysis showed that methemoglobin released iron ions into the retina and caused retinal lipid peroxidation.Fresh autologous blood was injected into the subretinal space of rabbits, which caused gradual PR degradation and edema.The iron in PRs and RPE was detected by Perls staining.The strongest mark was directly covered in the subretinal hemorrhage and disappeared farther from the blood accumulation point[63,82].According to electroretinogram measurements, the iron chelator desferrilamine was shown to protect these rabbits[82].The analysis of water samples collected from healthy people and patients with RVO showed that both VEGF-A and placental growth factor were significantly increased and significantly positively correlated with the increase and ischemic level[83].The main features of RVO include increased vascular permeability and continuous retinal ischemia; the more obvious retinal ischemia, the earlier neovascular occurs.The main features of RVO include increased vascular permeability and continuous retinal ischemia, wherein the more obvious the retinal ischemia,the earlier that neovascular occurs[84].In particular, the ischemic CRVO has a sudden and large amount of bleeding,which causes serious damage to the retina.In particular, the ischemic CRVO has a sudden and large amount of bleeding,which causes serious damage to the retina[85].The possible mechanisms of visual impairment caused by subretinal hemorrhage include mechanical damage to PRs and RPE,separation of PRs and RPE, formation of the fibrovascular membrane, and iron toxicity[86].In BRB damage caused by RVO, iron may be one of the catalysts for oxidative stress and an inflammatory response that may be connected to VEGF production.
Retinopathy of PrematurityRetinopathy of prematurity(ROP) has emerged as a major pathogenesis of loss of vision in kids around the globe[87].Under normal circumstances,peripheral retinal vascularization will continue to develop before the fetus approaches term.ROP develops in two different stages, namely the vascular attenuation phase(phase I) and the fibrovascular proliferative phase (phase II).In both instances, VEGF performs a key role, but with contrasting effects[88].In hyperoxic phase I, VEGF is suppressed, preventing normal retinal vascularization and causes some growing vessels to disappear.In the late stage of retinal development, oxygen demand increases, and hypoxia occurs.The outer boundary between vascularized and nonvascularized retinas develops retinal neovascularization,when VEGF is increased in this anoxic state[89].The severity of vascular occlusion in the first stage affects the subsequent neovascularization to a large extent.Note that in the first stage,a large amount of ROS will be produced, which will react with lipids and cause lipid peroxidation and deoxyribonucleic acid damage.In addition, due to the low content of antioxidants and chelatase in the retina at birth[89], premature infants are more vulnerable to ROS-mediated injury, which suggests that oxidative stress-induced injury may be a new and promising therapeutic target for ROP treatment[88,90].Genes involved in iron metabolism are highly enriched under hypoxic conditions,according to global gene expression profiling of human fetus retinal microvascular endothelial cells (RMECs) culturedin vitro.This suggests that oxidative damage mediated by the dysregulation of genes involved in iron homeostasis may contribute to ROP[91].Clinical studies have shown that transfusion iron loading is one of the risks of ROP in infants with a birth weight below 1250 g[92], and that transfusion 10d after birth is related to a nearly fourfold increased risk of severe ROP[93].Increased Fe2+in retinal vascular endothelial cells and mitochondria and increase in ROS levelswere detected in the ROP mouse model, which led to lipid peroxidation.Elabela significantly ameliorated mitochondria-dependent ferroptosis by mediating the cystine/glutamate antiporter [system x(c-)]/GPX4 axis, revealing that iron overload and iron death may play important roles in the early phase of ROP[94].
Age-related Macular DegenerationAge-related macular degeneration (AMD) is a retinal degenerative disease and the main cause of blindness in the elderly aged 65 years and older in industrialized nations[95-96].The macula’s pathological aging can result in two forms of AMD: dry or non-neovascular AMD and wet or neovascular AMD.The pathogenesis of AMD mainly involves oxidative stress and inflammatory responses leading to retinal PR death, and AMD is characterized by iron accumulation in the RPE that may be associated with the induction of oxidative stress and inflammatory responses[97].RPE causes iron to accumulate in RPE by phagocytosis of outer discs rich in iron and PUFAs[98].The residues of phagocytic PRs will accumulate with age, forming lipofuscin particles with phototoxicity in RPE, which containcarboxyethylpyrrole (CEP) and n-retinol-n-retinolamine (A2E)[99].CEP and A2E eventually become part of drusen, which can induce inflammation, including leukocyte hyperplasia.These inflammatory responses may reduce choroidal blood flow,hinder the diffusion of metabolic substances, and produce cytotoxicity, ultimately leading to degenerative changes in PRs and RPE cells[100].Polymorphisms in several genes involved in iron metabolism have been associated with risk factors for AMD, including TFR1, TFR2 (obesity, tobacco)[101],Dmt1[102], IRP1 and IRP2[102], and heme oxygenases 1 and 2(HO1/2)[103].A previous study found that ferritin light chain 1 mRNA expression significantly increased with age in the retina, whereas no significant changes in ferritin heavy chain 1 mRNA expression were observed.The increase in iron content in the retina with age is localized and does not involve changes in blood iron levels[104].High levels of iron have been found in the RPE, outer retina, and choroid among the elderly[105].The elderly have a 3-fold increase in RPE iron content and a 1.3-fold increase in the neural retina compared to young individuals[13].Iron accumulated in the RPE may contribute to the upregulation of VEGF production through the RPE by inducing oxidative stress and stimulating inflammation, thereby promoting the proliferation of aberrant blood vessels in the choroid[106-107].Bothin vitroandin vivo, ferroptosis was induced by light exposure in PR cells.Ferroptosis is characterized by elevated iron levels,mitochondrial contraction, glutathione consumption, elevated malondialdehyde (MDA), and reduced expression of the solute carrier family 7 member 11 and GPX4 proteins.PR cells expressed typical features of ferroptosis after exposure to light bothin vitroandin vivo, including increased iron levels, mitochondrial contraction, glutathione consumption,and malondialdehyde, as well as decreased solute carrier family 7 member 11 and GPX4 protein expression[8].The accumulation of iron caused by iron metabolism disorders can be distinguished from other oxidative stress pathways and has become a unique mechanism for inducing AMD.

Table 1 Comparison of three iron chelating agents used in clinical practice
IRON CHELATION THERAPY
Iron chelators were first used to treat patients with thalassemia who required repeated transfusions to cause systemic iron overload[108].Given that iron chelators have antioxidant and anti-inflammatory effects, in recent years they have been found to present potential therapeutic effects in various nonsystemic iron overload diseases.Apart from cancer[109]and neurodegenerative and IR injury[110], acquired immunodeficiency syndrome (ΑIDS) treatment[111]and corona virus disease 2019 (COVID-19) therapy[112]are also included in the current research on iron chelators.High intracellular iron levels are necessary for ferroptosis, which may be prevented by iron chelators, whether it is brought on by cys2 deprivation,system XC-inhibition, or direct GPX4 inhibition[13].
Clinically Used Fe ChelatorsAt present, the FDA has approved clinically available iron chelates, including deferoxamine,deferoprone, and deferasirox[29](Table 1)[2,12,17,113-115].
DesferrioxamineDesferrioxamine is a bacterial siderophore produced byStreptomyces muciniphila[113].It is a hydrophilic hexadentate iron chelator with a short biological halflife, a large molecular weight, and difficulty permeating through cell membranes, thus binding iron mainly in the blood and eliminating the formed chelate through urin[17,113].Deferoxamine has shown significant neuroprotective effects in animal models of intracerebral hemorrhage by forming stable complexes with hemosiderin, preventing iron entry into the Haber-Weiss reaction, and inhibiting oxidative stress,inflammation, phagocytosis, and apoptosis[116].Deferoxamine attenuates damage to RGCs and optic nerve fibers in mice with chronic ocular hypertension[115].Desferrioxamine alleviates iron death in RPE induced by oxidative stress[11],while desferriamine regulation of the HIF-1α/VEGF-Α pathway improves retinal hypoxia in rats with subarachnoid hemorrhage[19,117].
DeferiproneDeferiprone is a synthetic bidentate hydroxypyridinone Fe chelator that forms a neutral 3:1 chelator:Fe3+complex and is orally active.Owing to its low molecular weight and lipophilic nature, it is able to cross cell membranes and, therefore, chelate iron from cells.Deferiprone can permeate the blood-brain barrier, and it has also been shown to reduce labile iron in the retina[115], without causing retinal toxicity in mice[118].The activity of HIF prolylhydroxylases can be reduced and the antihypoxic response increased by removing or displacing iron, thus improving the hypoxic state in patients with COVID-19[119].
DeferasiroxDeferasirox,a tridentate chelator with high affinity for Fe3+, binds to iron in a 2:1 ratio to form a complex with a long half-life of about 8-16h.Deferasirox was approved by the US FDΑ in 2005 and is the first drug used orally to treat iron overload caused by blood transfusion[114].Deferasirox has the potential to be studied as a new drug to treat intracerebral hemorrhage, which can alleviate brain damage in ischemic stroke mouse models by chelating iron[120].Deferasirox can also affect nuclear factor NF-κB activity, as adding iron to cells does not affect the inhibition of NF-κB, which is an independent effect[121].Deferasirox combined with sorafenibinduced programmed cell death enhances the anti-tumor effect against hepatocellular carcinoma[109]and inhibits the NF-κB/HIF1α approach in leukemia[122].
Experimental Fe Chelators
DIBIDIBI is a modified hydroxypyridinone 3-hydroxy-1-(βmethacrylamidoethyl)-2-methyl-1(1 H)-pyridinonepolymer with a relatively low molecular weight (9 kDa), which binds Fe3+with greater selectivity than deferiprone[123].In an experimental acute lung injury, DIBI showed a powerful antiinflammatory effect[124].
BHAPIBHAPI ((N’-(1-(2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyloxy)phenyl)ethylidene) is a derivate of the salicylaldehyde isonicotinoyl hydrazone analogue HAPI(N’-[1-(2-hydroxyphenyl)ethyliden]isonicotinoylhydrazide)boronate-masked iron pro-chelator that has no chelating ability by itself but chelates iron only after the oxidative cell environment eliminates the boron-based mask from transitioning to HAPI[125].BHAPIviathe Wnt-β/Catenin pathway, attenuates oxidative stress-protective HT22 cells induced with glutamate[126].The coincubation of BHAPI with 10 mm paraquat-treated ARPE-19 cells for 48h mitigated oxidative stress, improving cell viability, whereas cells not treated with BHAPI died completely[125].HAPI treatment resulted in a fourfold increase in TFR mRNA levels compared to BHAPI, suggesting that HAPI caused low intracellular iron,whereas BHAPI did not significantly alter these levels.The advantage of pro-iron chelators is a small disturbance of body iron homeostasis that does not cause iron deficiency[127].
PhytochelatorsThere is potential for the development of several kinds of phytochelators and plant polyphenols that are comparable to chelating medications for therapeutic application.Mimosine and tropolone, two phytochelators,have been found to be orally active and efficient in animal models[128].Green tea extract regulates lipid peroxidation by chelating Fe2+and inhibiting β-oxidative tissue damage in thalassemic mice.Curcumin removes the LIP in hepatocytes and cardiomyocytes[129].Maltol, mimosine, morin, tropolone,and esculetin have been proven to have the antioxidant effect of chelated iron[130].
CONCLUSIONS AND PROSPECTS
Despite the indispensable physiological significance of iron inside the human body, an excessive accumulation of iron has the potential to inflict harm on the retina.Consequently, the utilisation of chelating agents presents itself as a promising therapeutic or supplementary strategy for addressing this issue.The current application of iron chelators to treat retinal vascular diseases is still in the basic research stage, and there are many unsolved challenges, such as the disruption of systemic or local iron homeostasis, the stability of chelates within cells, and so on.Ideally, the chelator of choice would only bind iron but not other divalent metals of great biological importance, such as zinc (Zn2+).Additionally, iron chelators must be able to successfully cross the BRB.Research on iron chelation in the therapy of retinal vascular disease is notably lacking, thus more has to be learned about how iron chelators work in order to create safer medications or chelation techniques for the aim of safe iron reduction.
ACKNOWLEDGEMENTS
Foundations:Supported by Luzhou Municipal People’s Government and Southwest Medical University(No.2021LZXNYD-J03); Science and Technology Department of Sichuan Province Project (No.2022YFS0611).
Conflicts of Interest: Li Y,None;Cheng ZX,None;Luo T,None;Lyu HB,None.
 International Journal of Ophthalmology2023年11期
International Journal of Ophthalmology2023年11期
- International Journal of Ophthalmology的其它文章
- Research progress on animal models of corneal epithelial-stromal injury
- Role of lymphotoxin alpha as a new molecular biomarker in revolutionizing tear diagnostic testing for dry eye disease
- Axial length and anterior chamber indices in elderly population: Tehran Geriatric Eye Study
- Development of a new 17-item Asthenopia Survey Questionnaire using Rasch analysis
- Retinal thickness and fundus blood flow density changes in chest pain subjects with dyslipidemia
- Analysis of independent risk factors for acute acquired comitant esotropia