
壳聚糖接枝PGMA增容PLA/PBAT复合材料的结构与性能
2023-11-08 15:29朱晔张鑫杨海存何明阳
工程塑料应用 2023年10期
朱晔,张鑫,杨海存,何明阳
(1.常州大学石油化工学院,江苏常州 213164; 2.常州大学生物质高效炼制及高质化利用国家地方联合工程研究中心,江苏常州 213164;3.常州大学材料科学与工程学院材料科学与工程国家级实验教学示范中心,江苏常州 213164)
石油基高分子材料的广泛使用及其不可生物降解的缺陷导致的日益严重白色污染使得具有可再生和可降解特性的高分子材料成为近二十年来的研究热点[1-2]。诸如聚羟基丁酸酯(PHB)、聚羟基丁酸酯-共戊酸酯(PHBV)、聚丁二酸丁二酯(PBS)和聚乳酸(PLA)等可生物降解高分子材料的合成工艺和改性方法已经广泛研究和发展,其中,PLA 是目前商业化程度最高、消费量最大的可完全生物降解和再生高分子材料,具有理想的生物相容性、强度、加工性和价格优势[3]。然而,在取代石油基非生物降解塑料(如聚丙烯、聚对苯二甲酸乙二酯等)方面,PLA 固有的高脆性和低断裂伸长率严重限制了其在柔性包装材料中的应用,就现有的PLA生产工艺而言,共混改性相较于化学共聚具有明显的优势和实际意义[4-5]。含柔性链聚合物,如聚烯烃、聚烯烃共聚物、柔性聚酯或共聚酯能有效提升PLA 的韧性,与聚烯烃类或其它可生物降解增韧组分相比,聚(对苯二甲酸-己二酸丁二酯)(PBAT)是一种价格便宜且具有生物降解性能的共聚酯,在增韧PLA的同时维持了其本身的可生物降解性[6-7]。然而,PLA与PBAT属于热力学不相容体系,仅在PBAT含量较少时二者为部分相容体系,过量的PBAT 会导致共混物出现明显的相分离,无法发挥增韧作用。
到目前为止,PLA/PBAT 共混体系常用增容方法是加入反应性增容剂,如环氧、酸酐、异氰酸酯等扩链剂,或反应活性较高的第三组分,其原理是在共混过程中原位形成不同的共聚物起到增容作用[8-10]。此外,在热力学不相容共混体系中加入粒子第三相也能起到增容作用,然而,这与粒子在体系中的分散状态密切相关[11-12]。当粒子处于单一相中,可改变相组成并诱导分散相纤维化,通过降低分散相尺寸起到一定的增容作用;当粒子处于两相界面时,可降低两相界面张力,从而降低分散相尺寸提供相容性,但粒子更倾向于分散在与其亲和力更强的聚合物相中。壳聚糖(CS)是一种无毒、可生物降解、生物相容的阳离子多糖,由甲壳素部分脱乙酰制得[13]。一方面,CS 具有良好的生物相容性、生物降解性和多官能团,是可生物降解高分子材料共混改性的常用组分,在共混体系中CS 能起到增强作用,同时解决了其加工性能差的缺陷[14];另一方面,CS 分子结构中含有大量可反应的羟基和氨基,这为通过化学改性调控CS 在PLA/PBAT 共混体系中的分散提供了重要基础[15]。
由于CS 分子间作用力较强,熔融温度高于分解温度,因此笔者通过离子交联法降低CS 颗粒分散尺寸,再经表面引发接枝聚合,制得CS接枝聚甲基丙烯酸缩水甘油酯(PGMA) (CS-PGMA),从而在CS中引入反应性增容基团,最后通过熔融共混法制备PLA/PBAT/CS-PGMA 复合材料,考察了CSPGMA的加入对于复合材料结晶行为、力学性能及界面相容性的影响。
1 实验部分
1.1 主要原料
PLA:4032D,美国Nature Works公司;PBAT:TH801T,新疆蓝山屯河聚酯有限公司;CS:脱乙酰度≥95%,黏度100~200 mPa·s,上海麦克林生化科技有限公司;
聚磷酸钠:分析纯,98%,上海阿拉丁生化科技股份有限公司;
四氢呋喃(THF)、甲基丙烯酸缩水甘油酯(GMA):分析纯,上海凌峰化学试剂有限公司;
硝酸铈铵(CAN):分析纯,上海阿拉丁生化科技股份有限公司;
硝酸:65%~68%,国药化学试剂有限公司;
乙酸:分析纯,上海阿拉丁生化科技股份有限公司。
1.2 主要仪器与设备
超声波清洗机:SB3200DT型,中国宁波新芝生物科技股份有限公司;
高速冷冻离心机:himac CR22G型,日本Hitachi公司;
冷冻干燥机:ALPHA 1-2 LD plus 型,德国Christ公司;
哈克微量成型系统:HAAKE PolyLab OS型,美国赛默飞世尔科技公司;
场发射扫描电子显微镜(FESEM):Supra55 型,德国蔡司公司;
差示扫描量热(DSC)分析仪:DSC8000型,美国Perkin Elmer公司;
动态热机械分析(DMA)仪:DMA8000 型,美国Perkin Elmer公司;
热重(TG)分析仪:TGA TG209 F3 型,德国耐驰公司;
傅里叶变换红外光谱(FTIR)仪:spotlight 200i型,美国Perkin Elmer公司;
电子万能试验机:UTM4103 型,深圳三思纵横科技股份有限公司。
1.3 样品制备
CS 交联纳米粒子的制备:将CS 溶解于质量浓度为1%的乙酸中,制备成3 mg/mL 的CS 溶液,然后向上述溶液中加入0.8 mg/mL 的三聚磷酸钠溶液,控制壳CS 与三聚磷酸钠的质量比为3∶1,室温搅拌1 h,随后离心3~4 次,将沉淀物放置于-80 ℃冰箱冷冻2 h 后再冷冻干燥36 h 得到CS 交联纳米粒子。
CS-PGMA 的制备:在250 mL 的单口烧瓶中将10 g CS交联纳米粒子超声分散在200 mL去离子水中,室温下缓慢通入氮气鼓泡1 h 后加入15 g 单体GMA 并持续搅拌30 min,随后加入5 mL 溶有0.5 g CAN 的硝酸溶液(0.5 mol/L),将烧瓶密封后置于50 ℃的恒温油浴锅中持续搅拌反应5 h。反应结束后抽滤分离出固体产物,用THF 洗涤数次,最后用去离子水反复洗涤至中性,冷冻干燥得到CSPGMA改性纳米粒子。
PLA/PBAT/CS-PGMA 复合材料的制备:将PLA和PBAT在80 ℃下真空干燥过夜,然后以质量比1∶1 的PLA/PBAT 为基体,采用哈克微量成型系统将PLA/PBAT 与不同比例的CS-PGMA 进行熔融共混,共混温度为180 ℃,螺杆转速为100 r/min,最后将复合材料注塑成拉伸性能测试样条,注塑温度为190 ℃。其中,CS-PGMA的质量分数分别为0%,1%,2%,4%和8%。
1.4 测试与表征
FTIR 分析:采用FTIR 法对复合材料中的成分进行定性测定。测量波数范围为4 000~500 cm-1,扫描次数为64次,分辨率为4 cm-1。
拉伸性能测试:利用电子万能试验机在室温条件下,参照GB/T 1040.3-2006,采用1B 型试样进行拉伸性能测试,试样标距为70 mm,拉伸速率为2 mm/min,每组试样测试3次取平均值。
DSC分析:采用DSC对PLA及其复合材料的结晶行为进行测试;取试样8~12 mg,氮气保护环境下由30 ℃以10 ℃/min的速度升温至180 ℃并记录熔融吸热曲线,得到PLA复合材料的结晶度和熔融温度,其中结晶度由式(1)计算。
式中:Xc为结晶度,单位%;ΔHm为熔融焓,单位J/g;ΔHc为冷结晶焓,单位J/g;ΔH0为PLA 完全结晶或熔融时的热焓,其值为93 J/g[16];ϕ为CS-PGMA和PBAT的质量分数。
DMA 分析:三点弯曲模式,样条尺寸为35 mm×12 mm × 3 mm,测试温度范围-60~100 ℃,频率为1 Hz,应变为0.1%,升温速率为10 ℃/min。
TG 分析:样品质量约为5 mg,氮气氛围下以10 ℃/min从室温升温至700 ℃。
FESEM分析:使用液氮脆断PLA/PBAT复合材料,采用FESEM观察断面形貌,加速电压5 kV。同时采用FESEM观察CS和CS-PGMA纳米粒子的微观形貌,加速电压5 kV。
降解试验:取适量室外土壤,将干燥至恒重的足量复合材料块状样品置于土壤中,初始质量为m1,定期加入适量自来水保持湿度,每隔一周取出5个样品,洗干净后充分干燥并称重,取平均值,记为m2,降解率Dr通过式(2)计算。
2 结果与讨论
2.1 CS和CS-PGMA的FTIR分析
图1为CS和CS-PGMA纳米粒子的FTIR谱图。图1中,CS纳米粒子谱图中3 424 cm-1处为O—H和N—H的伸缩振动峰,2 920,2 876 cm-1处为CS骨架中C—H 的反对称和对称伸缩振动峰,1 653,1 601 cm-1处为N—H 的反对称和对称弯曲振动峰,1 387 cm-1附近为骨架中C—H 弯曲振动峰,1 158 cm-1处为P=O伸缩振动峰,1 100 cm-1和1 032 cm-1处分别为C—N和C—O伸缩振动峰。经接枝聚合改性后,CS-PGMA 的谱图中在3 500 cm-1附近与羟基和氨基相关的特征峰强度明显减弱,这是因为接枝聚合的引发过程消耗了部分羟基和氨基。同时,在1 732 cm-1处出现了较强的PGMA 分子链中的酯基C=O伸缩振动峰,1 410 cm-1附近与C—H相关的特征峰强度明显增大,此外,受脂环醚基团的影响,1 060 cm-1附近的C—O,C—O—C 和C—N 的特征峰发生叠加而宽化。以上数据的变化证明通过接枝聚合成功制备了CS-PGMA改性纳米粒子。
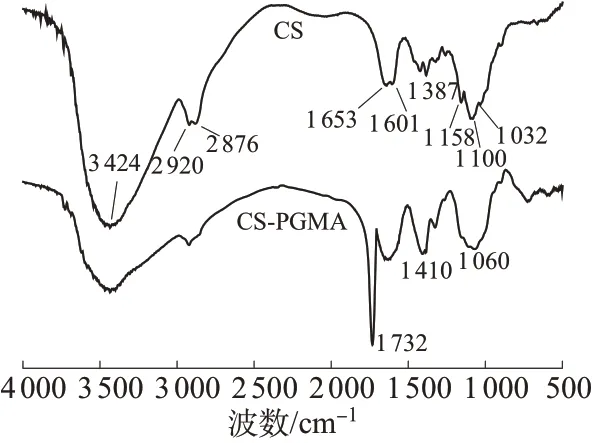
图1 CS和CS-PGMA纳米粒子的FTIR谱图
2.2 CS和CS-PGMA的FESEM分析
图2为CS和CS-PGMA纳米粒子FESEM照片。从图2a可以看出,由CS溶液经离子交联制备的CS纳米粒子尺寸分布在70~100 nm,且团聚非常严重,这可能是多个纳米粒子由于羟基和氨基之间的氢键作用导致的。经接枝聚合后,图2b 中CS-PGMA纳米粒子的分散得到显著改善,证明表面引发接枝聚合能显著降低粒子的分布尺寸,主要是由于接枝的PGMA 降低了粒子间的作用力,有效缓解了团聚,同时接枝聚合消耗了大量羟基和氨基,降低了分子间氢键的作用。单颗CS-PGMA纳米粒子的尺寸约为100~130 nm,相对纯CS纳米粒子有所增大,进一步证明发生了接枝聚合,但由于常规自由基的特点,接枝PGMA分子链的长度以及在干燥过程中的堆砌状态有所不同,使得CS-PGMA 纳米粒子微观形貌不规则。

图2 CS和CS-PGMA粒子的FESEM照片
2.3 CS-PGMA含量对复合材料力学性能的影响
图3为不同CS-PGMA 含量的复合材料的拉伸应力-应变曲线,拉伸强度和断裂伸长率列于表1。由图3和表1看出,不添加CS-PGMA时,PLA/PBAT共混物拉伸强度仅为29.12 MPa,断裂伸长率为83.84%,虽然PLA与PBAT均为聚酯,但是在热力学上为不相容体系,简单共混时的相容性较差。加入CS-PGMA 后,体系相容性得到提升,当CS-PGAM质量分数为4%时,拉伸强度达到最大值,为47.85 MPa,断裂伸长率为225.03%,表明CS-PGMA 的加入使得体系的强度和韧性趋向于平衡,PBAT 发挥了增韧作用,CS-PGMA起到了增强作用,基本能满足柔性包装材料的应用要求。CS-PGMA中接枝链含有大量环氧侧基,在熔融共混过程中可以和PLA及PBAT 发生酯化反应原位形成PLA-CS-PBAT 共聚物[17],从而起到反应增容作用,使得体系强度升高,且PBAT的增韧作用更加明显。增大CS-PGMA质量分数至8%时,CS-PGMA本身团聚的发生导致体系力学性能反而出现下降,上述力学性能的变化与体系微观结构密切相关。图4 为CS-PGMA 质量分数为0%和4%的复合材料断面形貌FESEM 照片。从图4a可以观察到明显的界面脱黏,进一步说明PLA 和PBAT 两相之间作用较弱。从图4b 可以看出,CS-PGMA 通过酯化反应提升了体系的相容性,两相界面相对于未加CS-PGMA 的共混物变得模糊。
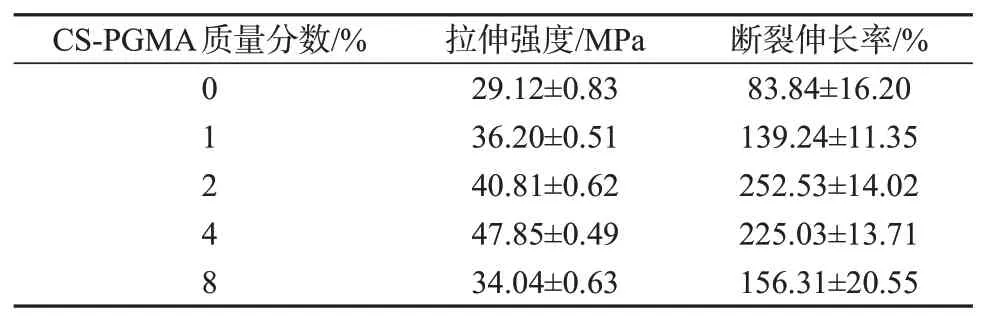
表1 不同CS-PGMA含量的复合材料力学性能
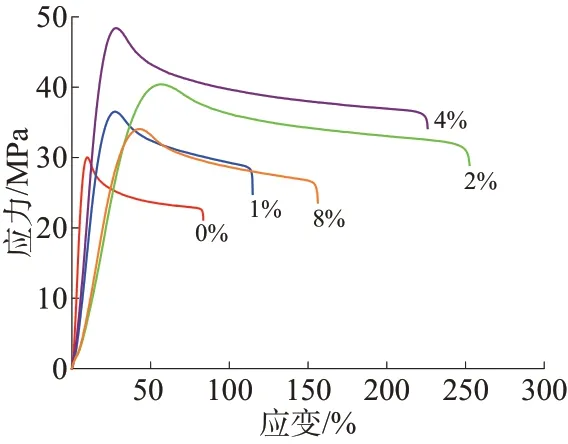
图3 不同CS-PGMA质量分数的复合材料的拉伸应力-应变曲线
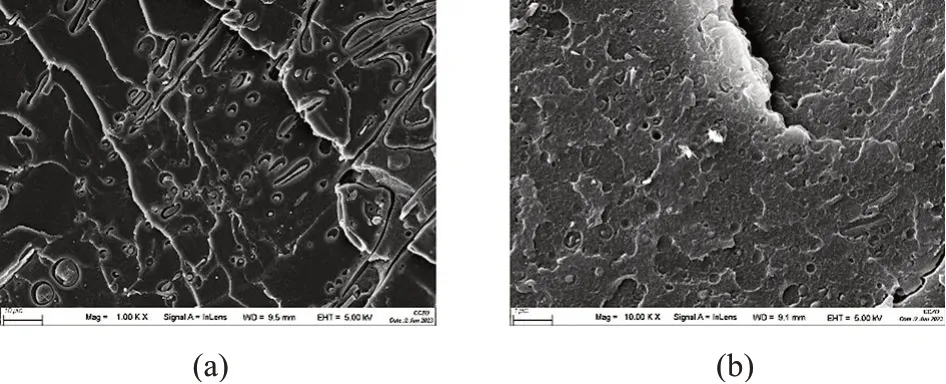
图4 CS-PGMA质量分数为0%和4%的复合材料断面形貌FESEM照片
2.4 CS-PGMA 含量对复合材料熔融行为和结晶的影响
不同CS-PGMA含量的复合材料的DSC熔融曲线如图5所示,由曲线得到的热力学参数见表2。由图5 和表2 可以看出,不含CS-PGMA 的PLA/PBAT共混物在84.2 ℃附近出现了峰较宽的冷结晶峰,且在154.2和161.8 ℃处出现了熔融双峰,其中温度较低的熔融峰为PLA冷结晶形成晶区的熔融,这部分晶区完善程度较低。随着CS-PGMA 含量的增加(质量分数未高于2%时),复合材料中PLA的冷结晶峰逐渐减弱,且向低温方向移动。这是由于PBAT的玻璃化转变温度(Tg)较低,加入CS-PGMA后体系相容性提升,运动能力更强的PBAT 相能提升周围PLA分子链段的运动能力,在较低的温度下就能排列进入晶格,此外,少量CS-PGMA对PLA冷结晶具有异相成核作用,这表明CS-PGMA质量分数在2%以内时能促进升温过程中PLA 发生冷结晶[18]。同时,熔融双峰特征逐渐转化为肩冲(CS-PGMA 质量分数为1%),直至呈现熔融单峰(CS-PGMA 质量分数分别为2%和4%),证明CS-PGMA 在促进冷结晶的同时可以提高结晶的完善性。当CS-PGMA含量继续增大时,冷结晶峰强度逐渐减小,这是由于增大CS-PGMA 含量能有效提升体系相容性,受反应性增容和PBAT 相细化的影响,PLA 在受限空间中的冷结晶活化能增大,整个升温过程中大部分PLA相来不及进行冷结晶,熔融曲线中仅能观察到一段较宽的微弱凸起。当CS-PGMA质量分数为8%时,熔融峰又出现肩冲,这可能是由于高含量的CSPGMA 纳米粒子发生明显团聚,且分布不均匀,导致结晶过程中形成的晶区完善程度差异增大,进而再次形成熔融峰肩冲现象。CS-PGMA表面羟基与PLA 和PBAT 之间发生化学反应形成共聚物,增大分子量的同时限制了分子链的有序排列,导致结晶度下降。然而,高含量的CS-PGMA粒子团聚严重,对体系结晶过程的影响下降,因此,结晶度随CSPGMA含量增大总体呈先下降后上升趋势。
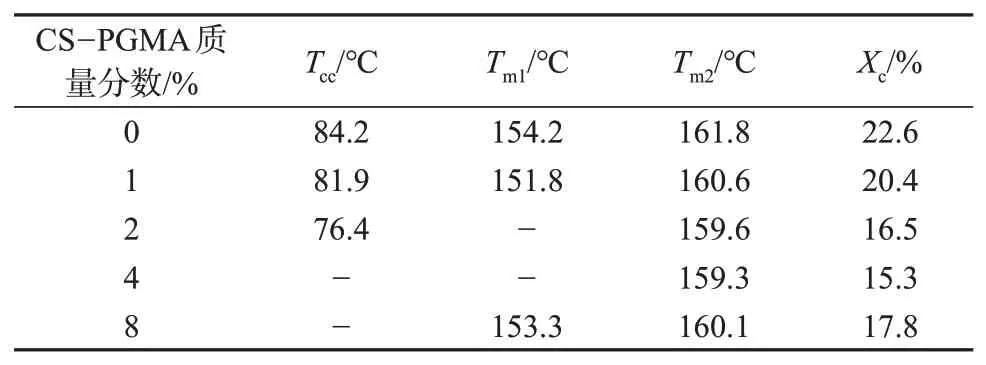
表2 不同CS-PGMA含量的复合材料的DSC参数
2.5 CS-PGMA含量对复合材料界面相容性的影响
图6为不同CS-PGMA含量的复合材料的DMA曲线。由图6可见,不含CS-PGMA的PLA/PBAT共混物的曲线上出现两个内耗峰,-24.6 ℃和59.7 ℃分别对应于PBAT 和PLA 的Tg,表明二者为热力学不相容体系[19]。当加入质量分数为1% 的CSPGMA 后,PBAT 的Tg由-24.6 ℃增大至-20.8 ℃,同时PLA 的Tg下降至58.1 ℃,以上结果证明CSPGMA 提升了PLA 和PBAT 的相容性。继续增大CS-PGMA质量分数至2%和4%时,PBAT的玻璃化转变受到抑制,曲线上逐渐观察不到明显的Tg,而PLA 的Tg分别下降至57.4 ℃和57.1 ℃。当CSPGMA 质量分数为8% 时,PLA 的Tg又上升至59.4 ℃,这可能是由于大量团聚体的形成降低了CS-PGMA的增容效果。
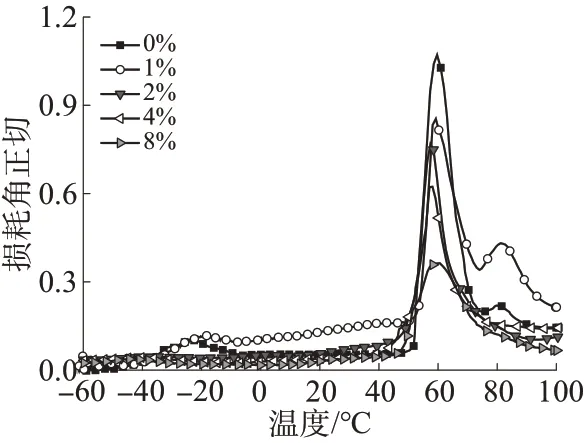
图6 不同CS-PGMA质量分数的复合材料的DMA曲线
2.6 CS-PGMA 含量对复合材料降解行为和热稳定性的影响
虽然PLA,PBAT 和CS 均属于可生物降解高分子,但降解速率有很大差别。图7为不同CS-PGMA含量的复合材料的降解曲线。由图7 可见,PLA 的降解速率较快,而PBAT 大分子结构中含有大量芳环,降解较慢,在不加CS-PGMA 时,共混物在42 d时的降解率为55.2%,明显低于文献中报道的纯PLA的降解率。虽然共混物为热力学不相容体系,结构缺陷较多,但PBAT 对整个体系的降解仍起到延迟作用。此外,整个降解过程中出现了明显的自加速现象,这可能是PLA的酸性降解产物一定程度上促进了PBAT的降解。随着CS-PGMA含量增大,复合材料的降解率出现了不同程度的下降,且自加速现象有所减弱。一方面,CS-PGMA 提高了PLA和PBAT的相容性,体系结构更加紧密,阻碍了土壤中的水和微生物进入材料内部,一定程度上阻碍了降解;另一方面,CS为碱性多糖,能中和一部分PLA的降解产物,推迟了自加速现象[20]。其中,CSPGMA 质量分数为4%的复合材料降解率最低,进一步增大CS-PGMA质量分数到8%时,降解率反而有所增大,这是因为CS-PGMA 团聚严重时易在材料表面产生缺陷,水和微生物能从缺陷处进入内部,更快地降解材料,从而使降解率增大。
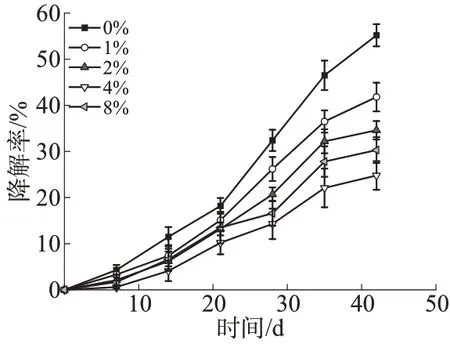
图7 不同CS-PGMA质量分数的复合材料的降解曲线
图8为不同CS-PGMA 含量的复合材料的TG曲线。由图8 可见,未加入CS-PGMA 时,共混物的TG曲线呈现出3个失重阶段:第1个失重阶段约为300~360 ℃,主要为PLA 的热分解;第2 阶段约为360~420 ℃,对应于PBAT 的热分解;第3 阶段的失重相对较为平稳,为前段分解产物的进一步分解,最终质量保持率接近0%。加入CS-PGMA后,复合材料在300 ℃之前出现了部分失重,这可能是由于CS和PGMA的起始分解温度均低于PLA和PBAT,CS-PGMA粒子中部分低分子量物质在升温过程中提前分解,导致体系结构变得疏松,使得部分PLA和PBAT发生了提前分解。此外,CS-PGMA分解产生的水和碱性产物对PLA和PBAT分解起到催化作用,导致复合材料的热稳定性随CS-PGMA 含量增大逐渐下降。

图8 不同CS-PGMA质量分数的复合材料的TG曲线
3 结论
(1)结合离子交联和表面引发接枝聚合成功制备改性纳米粒子CS-PGMA,改性纳米粒子的尺寸由CS 纳米粒子的70~100 nm 增大至100~130 nm,且团聚现象得到缓解,CS-PGMA 能有效增容PLA/PBAT热力学不相容体系。
(2)少量CS-PGMA 纳米粒子起到异相成核作用,促进冷结晶且提高结晶完善程度,随CS-PGMA含量增大,冷结晶逐渐受限,由熔融双峰向单峰转变,当CS-PGMA 质量分数为8%时,再次出现熔融峰肩冲现象。
(3)随CS-PGMA 含量增大,复合材料的结晶度和降解率均呈先下降后升高的趋势,热稳定性逐渐下降。
(4)当CS-PGMA质量分数为4%时,复合材料的拉伸强度达到最大,为47.85 MPa,断裂伸长率为225.03%,兼顾了材料的韧性和强度,有望作为高性能柔性包装材料使用。
猜你喜欢
学与玩(2022年12期)2023-01-11
合成树脂及塑料(2020年6期)2020-12-29
石油沥青(2019年4期)2019-09-02
中国塑料(2016年3期)2016-06-15
中国塑料(2016年9期)2016-06-13
中国塑料(2015年7期)2015-10-14
中国塑料(2015年1期)2015-10-14
新疆钢铁(2015年3期)2015-02-20
火炸药学报(2014年1期)2014-03-20
化学分析计量(2013年5期)2013-03-11
