
基于双向门控循环单元的5-甲基胞嘧啶位点预测
2023-11-07 09:15黄修威方中纯李海荣
中南民族大学学报(自然科学版) 2023年6期
黄修威,方中纯,李海荣
(1 内蒙古科技大学 信息工程学院,包头 014010;2 内蒙古科技大学 工程训练中心(创新创业教育学院),包头 014010)
迄今为止,已有170 多种化学修饰在RNA 中被发现[1-3].5-甲基胞嘧啶(m5C)是最常见的转录后修饰之一,它是通过RNA 甲基转移酶催化将甲基转移到胞嘧啶环的第5 位碳原子形成的.m5C 位点参与多种生物过程,包括调节蛋白质翻译和RNA-蛋白质相互作用,调节核mRNA 输出和RNA 可变剪接,增强RNA 稳定性等[4-7].此外,有研究还证明m5C 修饰与许多疾病有关,如乳腺癌[8]、常染色体隐性智力残疾[9]、肌萎缩侧索硬化[10]和帕金森氏病[11].因此,准确鉴定m5C 位点在RNA 中的位置是开展相应疾病和生物学功能研究的首要和关键任务.
目前已经开发出了数种方法来预测RNA 中的m5C位点.在实验中,已经开发了几种传统的高通量测序技术,如亚硫酸氢盐测序[12],m5C-RIP[13],mi-CLIP[14]和RBS-seq[15]等实验方法.虽然这些技术在鉴定不同物种的RNA 中的m5C 位点方面取得了一定的成功,然而,这些技术非常耗时和昂贵,难以跟上后基因组时代RNA 序列数量的急剧增加.因此又提出了多种基于机器学习算法来预测RNA 序列中m5C 位点的模型.FENG 等[16]首先在人类中通过伪二核苷酸组成(PseDNC)的三个理化特征设计了基于支持向量机(Support Vector Machine,SVM)的m5CPseDNC 模型.后来,QIU 等[17]开发了一个称为iRNAm5C-PseDNC 的模型,该模型建立在不平衡和冗余的数据集上并通过使用随机森林来预测人类的m5C位点.之后,FANG 等[18]开发了一个基于SVM的预测模型RNAm5CPred,来预测人类的m5C位点,并且使用独立的测试集来评估不同的方法.对于拟南芥中的m5C 位点,SONG 等[19]率先开发了预测模型PEA-M5C,在该模型中,结合了三种特征编码技术以提高综合性能.LI 等[20]从GEO 数据库中收集数据,基于随机森林算法开发了一个名为RNAm5 Cfinder 的模型,可用于预测小鼠和人类的m5C 位点.2020 年CHEN 等[21]通过引入位置特异性倾向相关特征,建立了一个新的模型m5CPred-SVM,来预测三种不同物种的RNA m5C 位点.同年DOU 等[22]提出了一种新的基于支持向量机(SVM)的工具,称为iRNAm5C_SVM,其通过组合多个序列特征来识别拟南芥中的m5C 位点.最近,CHAI 等[23]通过采用特征融合策略来利用信息序列,并堆叠集成学习框架,结合了五种流行的机器学习算法提出了Staem5模型,用于小鼠和拟南芥m5C位点的识别.
传统机器学习方法的性能高度依赖于特征提取的有效性,然而特定RNA 修饰最相关的特征组合很难判断.基于深度学习的计算架构能够在没有任何人工干预的情况下从序列中提取最重要的特征,并进行端到端预测.通过多个神经元层和激活函数,它将获得从原始输入到潜在表征的映射能力,而潜在表征是由标记数据训练的.在这样的数据驱动模型下,与RNA 修饰位点语义信息相关的RNA序列深层特征就会显现出来.
为了弥补现有计算模型在性能和计算成本方面的缺点,本文提出了一种简单有效的基于双向门控循环单元(BiGRU)的预测模型pm5C-BGRU,用于鉴定RNA 序列中的m5C 位点.输入RNA 序列由独热编码(One-hot encoding)和核苷酸化学性质(NCP)的组合来表示.核苷酸的化学性质是核苷酸在官能团、氢键和环结构方面最基本的表征.BiGRU能够从RNA 序列中学习序列前后特征间的长期依赖关系,这使得pm5C-BGRU 能够更准确地识别m5C 位点.最后使用10 倍交叉验证和独立数据集测试的方法来评估和比较pm5C-BGRU的性能.
1 数据和方法
1.1 数据集
高质量的数据集对于训练和评估预测模型都极其重要.从最近发表的文献中收集了三个物种的m5C 数据.拟南芥的m5C 数据从NCBI 基因表达综合(GEO)数据库获得,登录号为GSE94065[24].因为拟南芥的m5C 样本足够大,所以使用CD-HIT 程序[25]通过将序列同一性的阈值设置为80%来去除冗余样本.最终,在拟南芥基因组中获得了6289 个阳性样本.拟南芥的阴性样品从它们的基因组中获得,为了平衡阳性和阴性样本,随机挑选相同数量的拟南芥阴性样本并使用CD-HIT 程序与阳性样本进行相同操作.小鼠和人类数据集来自于CHEN等[21]的研究工作,其中分别包含小鼠和人类的5563和269 个阳性样本及相同数量的阴性样本.所用样本均已使用CD-HIT程序去除数据集中的相似序列.
基准数据集通常分为两部分,一个是训练数据集,另一个是独立测试集.训练数据集用于模型构建、交叉验证和确定学习算法的超参数.独立测试数据集用于测试性能和模型泛化能力.三个物种的训练和独立测试数据集数量如表1所示.

表1 不同物种的m5C数据集Tab.1 m5C data sets of different species
样本中每个RNA 序列是长度为41 bp 的RNA片段,这是现有工具中最常用的长度.m5C数据集中的每个RNA片段可以表示如下:
其中中心X 是靶向位点,表示m5C 的C(胞嘧啶核苷酸).N1到N20代表朝向靶位点的上游侧翼核苷酸,N22到N41而表示其下游侧翼核苷酸.
1.2 RNA片段的编码
pm5C-BGRU 采用RNA 序列作为输入.RNA 序列由独热编码和核苷酸化学性质(NCP)两种常用编码技术的组合来表示.独热编码是序列分析中最常见和最有效的编码方式之一,它将RNA 中的每种核糖核苷酸序列编码为一个独热向量,RNA 片段的A(腺嘌呤核苷酸)表示为[1,0,0,0],C(胞嘧啶核苷酸)表示为[0,1,0,0],G(鸟嘌呤核苷酸)表示为[0,0,1,0],U(尿嘧啶核苷酸)表示为[0,0,0,1].而NCP 是RNA 序列中每个核苷酸在三维笛卡尔坐标系中基于它们的三个化学基团的表示.RNA 序列中的四个核苷酸每一个都有不同的化学特性,如表2所示.考虑到环结构,A 和G 是由两个环组成的嘌呤,而C和U 是由一个环组成的嘧啶;就二级结构形成而言,A 和U 之间的键是弱氢键,而C 和G 之间的键是强氢键;关于化学官能团,A和C属于氨基,而G和U属于酮基.根据这三种化学性质,这四种核苷酸可以通过赋值1或0分为三个不同的组,即A表示为[1,1,1],C 表示为[0,1,0],G 表示为[1,0,0],U 表示为[0,0,1].通过将独热编码和NCP 产生的向量连接在一起,RNA 序列中每个核苷酸均被编码成一个7 维的特征向量,如图1 所示,即A=[1,0,0,0,1,1,1],C=[0,1,0,0,0,1,0],G=[0,0,1,0,1,0,0],U=[0,0,0,1,0,0,1],每个RNA 序列由41 个这样的列表组成,即一条RNA 片段可以编码成一个7×41的矩阵.

图1 RNA序列编码Fig.1 RNA sequence coding

表2 核苷酸化学性质Tab.2 Chemical properties of nucleotides
1.3 门控循环单元
循环神经网络(Recurrent Neural Network,RNN)是一种能够记忆上下文信息的网络架构,非常适合生物序列分析[26].门控循环单元(Gated Recurrent Unit,GRU)为RNN 的精简版,不但能记住上一时刻的输入信息,而且能够抑制梯度爆炸或梯度消失的现象.门控循环单元结构简单,只有两个门控单元,分别是重置门和更新门.重置门决定着上一时刻隐含层被遗忘信息的大小,更新门衡量过去信息的重要程度,决定着传递到当前隐含层信息的大小.GRU网络的基本结构如图2所示.
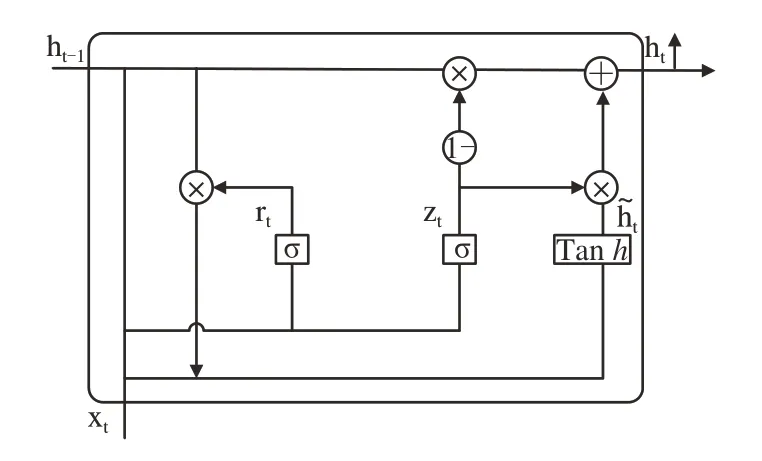
图2 GRU网络基本结构图Fig.2 Basic Structure of GRU Network
图2 中x1为模型输入,ht-1为上一时刻隐含状态,ht为当前时刻隐含状态,为候选隐含状态,rt代表重置门,zt代表更新门.具体计算过程如下:
式中,σ、Tanh为Sigmoid激活函数和Tanh激活函数;Wz、Wr、W为遗忘门、更新门、隐藏层对应的权值.
对于序列特征不明显的RNA 修饰位点预测问题,要考虑的不仅仅是在之前的信息,之后的信息也同样重要.双向门控循环单元(BiGRU)能够同时在发生位点前后考虑输入数据的特征,并集成它们,其结构如图3所示.

图3 BiGRU网络Fig.3 BiGRU network
BiGRU 网络多了一层隐藏层,信息可以通过正向和反向输入,第x个的输出为:
本研究中通过将RNA 序列信息输入到BiGRU网络,来找寻修饰位点周围核糖核苷酸排列的潜在规律.本模型pm5C-BGRU 搭建了两个尺寸为64 个神经元的BiGRU 网络层,在BiGRU 层之后,设置了一个尺寸为64 个神经元的全连接(fully connection)层.两层之间用一个 flatten 层来连接,同时为防止过拟合,在三个网络层的后面引入了一个参数为0.3的 dropout 层.所有网络层的激活函数均为ReLU 函数,它能够产生稀疏输出并加速收敛.优化器使用Adam 优化器,并且将学习率设置为2e-5.为了避免过拟合,设置early-stopping函数的规则是:当损失函数在300 次训练过程中都没有减小时,提前结束训练.网络结构具体参数如表3 所示,模型框架如图4所示.

图4 模型框架示意图Fig.4 Model framework

表3 BiGRU网络详细超参数Tab.3 BiGRU network structure and hyper-parameters
1.4 评价指标
为了测量模型的性能,本文计算了敏感性(Sn)、特异性(Sp)、准确性(Acc)和马修斯相关系数(MCC).这些性能指标用公式表示如下:
其中TP为真阳性,TN 为真阴性,FP为假阳性,FN 为假阴性.Sn 反映了被正确识别的真阳性类别的比例,Sp反映正确识别的真阴性类别的比例,Acc根据正确检测的正样本和负样本来呈现模型的总体性能.MCC 是一个比较均衡的指标,描述预测结果与实际结果之间的相关系数.此外,受试者工作特征曲线(ROC)的曲线下面积AUC 也是分类模型常见的一种性能度量,它表示不同类之间可分离的程度,可评估预测的整体性能.
1.5 实验环境
操作系统Windows10,CPU 型号为Intel®Core™i7-10700 CPU@2.90 GHz,运行内存为32 GB.该预测模型是基于Pytorch 深度学习框架,编程语言为Python.
2 结果与讨论
2.1 序列组成分析
为了更好地预测人类、小鼠和拟南芥的m5C 位点,使用Two Sample Logos[27]比较三个物种的m5C位点和非m5C 位点周围核苷酸的位置保守性,研究m5C 位点侧翼核苷酸的分布和偏好.三个物种序列位置保守的标志如图5所示.显然,就核苷酸分布而言,含m5C 的序列与非m5C 样品有着显著不同.小鼠序列中G 和C 在含有m5C 位点的序列中显著富集,而A和U在不含m5C的序列中显著富集.对于拟南芥,在m5C位点的上游序列有大量的C,相反嘌呤在非m5C上游序列中富集.另外,与拟南芥中C的高丰度相比,其他两个物种在含有m5C 位点的序列中有核苷酸G 的富集.图5表明,在m5C位点周围存在特定的核苷酸偏好,因此通过使用序列信息构建预测模型是合理的.此外,不同物种之间m5C 位点上下游核苷酸的差异说明应当构建物种特异性预测模型.

图5 m5C和非m5C位点周围的核苷酸分布图Fig.5 Nucleotide distribution around m5C and non m5C sites
2.2 不同编码方法的结果比较
在三个物种训练数据集上使用10 折交叉验证对所提出的模型进行评估.为了研究One-hot encoding 和NCP 整合后的有效性,进行了三组实验.第一组实验只使用了One-hot encoding 表示RNA 序列,第二组实验仅使用NCP 来表示RNA 序列,而第三组实验整合了这两种编码方法.这些实验的结果如表4所示.可以看出,与单独使用One-hot encoding或NCP 相比,将两种编码方法进行整合后的模型,在敏感性、特异性、准确性、MCC 方面都均有提升,可以在m5C位点的鉴定中产生更好的结果.

表4 不同编码方式下模型的性能比较Tab.4 Performance comparison of models under different coding methods
2.3 与现有预测工具对比
本节在独立测试集上将本文提出的pm5CBGRU 模型与近期已发表的预测模型进行了比较.因为不同预测方法使用了不同的基准数据集,所以使用独立测试集可以更客观.对人类用pm5C-BGRU与RNAm5CPred[18]和m5CPred-SVM[21]比较;小鼠和拟南芥则用pm5C-BGRU 则与m5CPred-SVM 和Staem5[23]进行比较.表5 显示了不同模型对相应物种的预测结果.

表5 pm5C-BGRU与现有方法的性能比较结果Tab.5 Performance comparison results of pm5C-BGRU and existing methods
从表5 可以看出,pm5C-BGRU 在人类的独立测试集上几乎在所有评估指标上都是最佳的.在小鼠的独立测试集上,Staem5 的Sp 要高于pm5C-BGRU,但其他评估指标还是pm5C-BGRU 最优.然而,在拟南芥的独立测试集上,m5CPred-SVM 与pm5CBGRU 取得了相近的预测性能,Acc 仅仅相差0.2.尽管两个模型在拟南芥独立测试集上的评价指标相似,但与m5CPred-SVM 相比,pm5C-BGRU 没有手工特征提取的过程,使得模型更加简便.所有这些结果表明,pm5C-BGRU 模型在预测m5C 位点方面比其他现有方法表现得更好.
2.4 跨物种预测
本研究分别为人类、小家鼠和拟南芥三个物种建立了三个模型.为了评估这些模型的物种特异性和可移植性,本节通过使用其中任一模型来测试该模型对其他物种中的m5C 位点的识别性能,测试结果如图6所示,其中横坐标为相应模型.
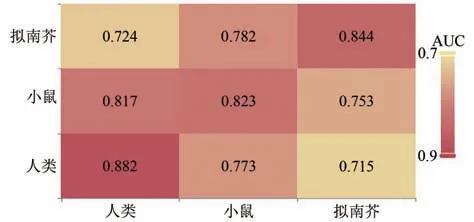
图6 跨物种预测性能Fig.6 Cross species prediction performance
由图6 可知,基于自身数据构建的模型在自己的独立测试集上表现良好,而在其他物种上的表现不理想.另外,人类与小鼠相互间的测试结果要优于人类与拟南芥、小鼠与拟南芥相互间的测试结果,这可能是因为人类和小鼠均为哺乳动物,亲缘关系较近,而拟南芥为植物的缘故.
3 结论与分析
本研究提出了基于深度学习框架的模型pm5CBGRU,用于准确地识别RNA 序列中的m5C 位点.pm5C-BGRU 通过利用独热编码和用于表示RNA 序列的核苷酸化学性质组合来提取重要的特征,这种组合有助于实现高效的m5C 位点鉴定结果.并且,与现有的机器学习预测模型相比,pm5C-BGRU表现出了更好的性能.该模型通过精确预测m5C 位点,将对m5C 的生物学功能和相关疾病的研究起到帮助.在后续的工作中,可以考虑将与特定疾病相关的m5C 位点使用深度学习方法进行预测,但扩展相关疾病的m5C数据集也是一项挑战.
猜你喜欢
中学生天地(A版)(2023年1期)2023-02-17
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
生物学通报(2019年3期)2019-02-17
生命科学研究(2018年1期)2018-05-29
上海农业学报(2017年3期)2017-04-10
山东农业工程学院学报(2016年6期)2016-12-01
