
超声波辅助H2O2-VC降解人参多糖对其结构特征与生物活性的影响
2023-11-07 11:08李福蕊高佳坤王丽岩刘学军
食品科学 2023年19期
王 尧,李福蕊,罗 源,高佳坤,王丽岩,刘学军
(吉林农业大学食品科学与工程学院,吉林 长春 130000)
多糖是分子质量在数万甚至数百万道尔顿的高分子聚合物,普遍存在于动物、植物和微生物中[1]。由于多糖来源广泛、副作用小,且具有抗肿瘤[2]、抗炎[3]、抗菌[4]、抗疲劳[5]、抗高血压[6]、降血糖[7]和免疫调节[8]等活性功能,越来越受到人们的关注。多糖的生物活性与其分子质量紧密相关,低分子质量的多糖具有更好的生物活性,因此其在开发功能性食品和医学应用时备受关注。
人参(Panax ginsengC.A.Mey.)又称黄精、地精、神草等,五加科人参属多年生草本植物,是一种药食同源的名贵药材[9]。人参中含有多种有益健康的活性成分,包括人参皂苷、肽聚糖、多糖、含氮化合物、脂肪酸和酚类化合物等[10]。近年来,从人参中提取的多糖已被证明具有降血糖活性,Liu Wei等[11]从党参残渣中提取纯化出一种中性多糖CERP1,对2型糖尿病(type 2 diabetes mellitus,T2DM)小鼠有明显的降血糖作用。然而,天然多糖的生物活性会受到其分子质量、结构特征和溶解度的影响[12]。比如,多糖较高的相对分子质量和较差的溶解度会限制其在生物体内发挥功能活性[13]。所以,有必要采用一种高效简便的降解方法来提高人参多糖(ginseng polysaccharides,GPS)的生物活性功能。
多糖的降解方法包括物理法、化学法和生物法。物理方法(如超声波、微波、辐射等)对设备要求较高,而且降解产物得率低。化学方法(如酸、碱水解等)因其水解条件苛刻可能导致糖苷键裂解不足。生物方法(如酶水解、微生物发酵等)因其特异性,需要多种酶或者微生物组合来降解多糖,导致降解成本相对较高,无法在食品工业中广泛应用[14]。近年来,H2O2因其可产生大量的氧自由基(如·OH)、对原料无污染、反应条件温和的特性引起了人们的注意[15]。同时,低浓度的抗坏血酸(VC)可以产生羟自由基[16],超声波可以通过空化作用使气泡大量形成和破裂,加速H2O2的分解,从而降解多糖[17]。目前关于超声波辅助H2O2-VC降解方法制备人参降解多糖的研究还鲜见报道。因此,本研究对GPS进行分离纯化,并利用超声波辅助H2O2-VC方法降解分离纯化获得的多糖组分GPS-1A,得到具有更高降血糖和抗氧化活性的低分子质量多糖DGPS-1A,进一步对其结构进行分析,为GPS的开发和利用提供参考。
1 材料与方法
1.1 材料与试剂
人参购买于吉林长春长白山地区,经鉴定为人工栽培长白山人参。人参经过清洗干燥后进行超微粉碎,过80 目筛,在95%(体积分数,下同)乙醇溶液中浸泡48 h后得到人参脱脂粉,烘干后于阴凉处保存备用。
苯酚、硫酸、葡萄糖、氯化钠、氯仿、正丁醇、VC、硫酸亚铁、H2O2、三氯乙酸 大连美仑生物技术有限公司;DEAE-纤维素-52、葡聚糖凝胶Sephadex G-100 上海阿拉丁生化科技股份有限公司;半乳糖醛酸、牛血清白蛋白 北京索莱宝科技有限公司;考马斯亮蓝 北京鼎国昌盛生物技术有限责任公司;溴化钾、1,1-二苯基-2-三硝基苯肼(1,1-diphenyl-2-picrylhydrazyl,DPPH)、水杨酸 上海麦克林生化科技有限公司。以上试剂均为分析纯。
1.2 仪器与设备
RHP-600高速多功能粉碎机 浙江荣浩工贸有限公司;HW.SY21-K电热恒温水浴锅 北京市长风仪器仪表公司;JY99-IIDN超声波细胞粉碎机 宁波新芝生物科技股份有限公司;1100高效液相色谱仪、IC-5000离子色谱仪 美国Agilent公司;层析柱(32 mm×30 cm;16 mm×60 cm)大连美仑生物技术有限公司;傅里叶变换红外光谱仪 美国Thermo Fisher公司;FD-80真空冷冻干燥机 北京博医康实验仪器有限公司;Mira4扫描电子显微镜 泰思肯(中国)有限公司。
1.3 方法
1.3.1 人参粗多糖的提取
参考李先华[18]的方法提取人参粗多糖。准确称取100 g除脂人参粉末,用蒸馏水在料液比1∶30、提取时长3 h的条件下重复提取两次,合并上清液,用旋转蒸发仪将多糖提取液浓缩至原体积的1/10,加入体积分数95%乙醇溶液过夜,5 000 r/min离心20 min,弃去上清液,沉淀用蒸馏水复溶后加入Sevag试剂(V(正丁醇)∶V(氯仿)=1∶4)进行脱蛋白处理,重复多次,直至无明显蛋白残留。将溶液在4 ℃下透析48 h,冷冻干燥,得到人参粗多糖,命名为GPS,多糖得率按公式(1)计算。
1.3.2 GPS的分离纯化
将GPS复溶后缓慢加入DEAE-纤维素-52层析柱(32 mm×30 cm),依次用0、0.1、0.3、0.5 mol/L NaCl溶液进行洗脱,流速为2 mL/min,每10 mL收集一管。将GPS-1(GPS的主要组分)合并、透析、浓缩和冻干。将GPS-1加入葡聚糖凝胶Sephadex G-100(16 mm×60 cm)柱,用蒸馏水以0.2 mL/min的流速洗脱,收集洗脱液,每5 mL一管,根据峰值合并得到分离组分,命名为GPS-1A,将其冷冻干燥后于4 ℃保存。
1.3.3 人参降解多糖的制备
采用超声波辅助H2O2-VC法对分离纯化得到的多糖GPS-1A进行降解,参考Xu Kaiqian等[19]的方法并稍作修改。根据本课题组前期得到的最佳工艺参数进行降解,配制4.2 mg/mL多糖溶液200 mL,加入10.3 mmol/L H2O2和10.3 mmol/L VC,在1 006 W超声功率下降解2 h。降解后调节溶液pH值至中性,去离子水透析48 h(1 000 Da透析袋),减压蒸馏浓缩后冻干得降解多糖,命名为DGPS-1A。
1.3.4 GPS的化学成分测定
通过苯酚-硫酸法[20]在490 nm波长处测定溶液中多糖的质量分数,以无水葡萄糖作为标准品。蛋白质量分数通过考马斯亮蓝法[21]测定,以牛血清白蛋白作为标准品。糖醛酸质量分数采用咔唑-硫酸法[22]测定,以D-半乳糖醛酸作为标准品。
1.3.5 GPS的结构表征
1.3.5.1 相对分子质量的测定
采用高效凝胶渗透色谱测定多糖的相对分子质量。将4.0 mg/mL样品溶解在蒸馏水中并使用0.45 µm滤膜过滤。每次运行注入10 µL样品溶液。流动相为0.05 mol/L Na2SO4溶液,流速为0.6 mL/min。使用不同分子质量的葡聚糖作为标准品。
1.3.5.2 单糖组成测定
通过1-苯基-3-甲基-5-吡唑啉酮(1,2-dihydro-5-methyl-2-phenyl-3H-pyrazol-3-one,PMP)衍生化和高效液相色谱(high performance liquid chromatography,HPLC)测定GPS和DGPS-1A的单糖组成。将5 mg样品在105 ℃用4 mL三氟乙酸(2 mol/L)水解6 h。将残留物重新溶解在4 mL甲醇中,反复干燥。将最终残留物溶解在50 mL超纯水中,使用0.22 μm滤膜过滤,并用离子色谱仪进行分析。
1.3.5.3 傅里叶变换红外光谱测定
将2 mg干燥样品与200 mg溴化钾粉末混合并压制成片,在4 000~500 cm-1的范围内进行傅里叶变换红外光谱扫描。
1.3.5.4 核磁共振氢谱测定
将25.0 mg多糖样品溶于0.5 mL D2O,并转移到核磁共振(nuclear magnetic resonance,NMR)管中。用光谱仪在室温下获得1D(1H和13C)NMR图谱。主要参数设置如下:脉冲序列zgpg30(1H和13C);延迟时间2 s,扫描频率4 000~30 000 Hz;扫描次数60~100 000 次;采集时间0.5~1.5 s;测试温度298 K。
1.3.5.5 扫描电子显微镜观察
将干燥后的多糖置于镀有金粉(100 nm厚)的铝板上。用扫描电子显微镜观察多糖的形态。
1.3.5.6 刚果红和I2-KI实验
将2.0 mL刚果红溶液(80 μmol/L)与2.0 mL多糖溶液(2 mg/mL)混合,加入一定量的氢氧化钠,使氢氧化钠的最终浓度在0~0.5 mol/L范围内,在室温下反应5 min,用紫外-可见分光光度计测定最大吸收波长(λmax)。
将8.0 mL I2-KI溶液(0.2%)与2.0 mL多糖溶液(2.0 mg/mL)混合,稀释10 倍体积后用紫外-可见分光光度计在190~400 nm波长范围内测定混合物的吸光度。
1.3.6 GPS体外降血糖活性的测定
1.3.6.1α-葡萄糖苷酶抑制率测定
将40 μLα-葡萄糖苷酶溶液(0.2 U/mL,pH 6.8)加入到不同质量浓度(0.2、0.4、0.6、0.8、1.0、2.0、4.0、6.0 mg/mL)40 μL多糖溶液中,在37 ℃下孵育5 min后加入20 μL对硝基苯-β-D-吡喃半乳糖苷(4-nitrophenylβ-D-glucopyranoside,PNPG)(0.5 mmol/L,pH 6.8),37 ℃恒温孵育30 min,加入150 μL碳酸钠溶液(0.2 mol/L)终止反应。在405 nm波长处测定吸光度,并用阿卡波糖作阳性对照。α-葡萄糖苷酶抑制率按公式(2)计算。
式中:A1为样品溶液+α-葡萄糖苷酶的吸光度;A0为样品溶液+0.1 mol/L pH 6.8磷酸盐缓冲液(phosphate buffer saline,PBS)的吸光度;A2为α-葡萄糖苷酶+0.1 mol/L pH 6.8 PBS的吸光度。
1.3.6.2α-淀粉酶抑制率测定
在1 mL多糖溶液(0.2、0.4、0.6、0.8、1.0、2.0、4.0、6.0 mg/mL)中加入1.0 mLα-淀粉酶溶液(0.5 mg/mL,pH 6.6),然后加入1.0 mL淀粉溶液(0.01 g/mL),在37 ℃下反应20 min,加入5 mL 3,5-二硝基水杨酸(3,5-dinitrosalicylic acid,DNS)试剂终止反应,沸水浴10 min。在540 nm波长处测定吸光度,以阿卡波糖为阳性对照。α-淀粉酶抑制率按公式(3)计算。
式中:A1为样品溶液+α-淀粉酶的吸光度;A0为样品溶液+0.1 mol/L pH 6.8 PBS的吸光度;A2为α-淀粉酶+0.1 mol/L pH 6.8 PBS的吸光度。
1.3.7 GPS体外抗氧化活性的测定
1.3.7.1 DPPH自由基清除能力测定
将2 mL多糖溶液(0.2、0.4、0.6、0.8、1.0、2.0、4.0、6.0 mg/mL)与1 mL DPPH溶液(0.1 mmol/L)混合,振荡均匀后在室温条件下黑暗中反应30 min。以VC作为阳性对照,在517 nm波长处测定溶液的吸光度。根据公式(4)计算DPPH自由基清除率。
式中:A1为样品溶液+DPPH溶液的吸光度;A0为样品溶液+无水乙醇的吸光度;A2为蒸馏水+DPPH溶液的吸光度。
1.3.7.2 羟自由基清除能力测定
将1 mL不同质量浓度(0.2、0.4、0.6、0.8、1.0、2.0、4.0、6.0 mg/mL)的多糖溶液分别与1 mL FeSO4溶液(9 mmol/L)和1 mL水杨酸溶液(9 mmol/L)振荡均匀,然后迅速加入1 mL H2O2(8.8 mmol/L),在37 ℃下水浴30 min。以VC为阳性对照,在510 nm波长处测定溶液的吸光度。羟自由基清除率按公式(5)计算。
式中:A1为样品溶液+H2O2溶液的吸光度;A0为样品溶液+0.1 mol/L pH 6.8 PBS的吸光度;A2为蒸馏水+H2O2的吸光度。
1.3.7.3 还原能力测定
将1 mL不同质量浓度(0.2、0.4、0.6、0.8、1.0、2.0、4.0、6.0 mg/mL)的多糖溶液分别与0.2 mol/L PBS(pH 6.6,2.5 mL)和2.5 mL铁氰化钾(质量分数1%)混合,在50 ℃下反应20 min,冷却至室温后加入2.5 mL三氯乙酸(0.1 g/mL)终止反应。将2.5 mL反应溶液与2.5 mL去离子水以及0.5 mL三氯化铁(0.001 g/mL)混合,在室温下反应10 min,以VC为阳性对照,在700 nm波长处测定溶液的吸光度。
1.4 数据处理与分析
所有实验均重复3 次,结果表示为平均值±标准偏差,使用SPSS 23.0软件对数据进行分析,采用单因素方差分析进行显著性分析(以P<0.05表示差异显著),采用GraphPad Prism 8软件作图。
2 结果与分析
2.1 GPS的分离纯化及降解结果
采用水提醇沉法提取人参粗多糖GPS,其得率为12.46%(基于原料干质量计算)。利用阴离子交换柱层析法对GPS进行分离纯化,其梯度洗脱如图1A所示。GPS经过分离纯化得到2 个多糖组分,分别为去离子水洗脱获得的中性多糖组分GPS-1和0.5 mol/L NaCl洗脱获得的酸性多糖组分GPS-2。收集样品,浓缩、透析、冻干后分别得到白色(GPS-1)和黄色(GPS-2)粉末。GPS-1和GPS-2的得率分别为52.14%和11.67%(基于GPS质量计算)。对初步纯化组分进行体外降血糖实验,结果显示两种多糖对α-葡萄糖苷酶的抑制率分别为45.56%和43.78%,无显著差异(P>0.05)。采用葡聚糖凝胶层析法对GPS-1进一步纯化,结果如图1B所示。GPS-1经Sephadex G-100分离得到一个相对均一的洗脱峰,经浓缩、透析、冻干后得到白色粉末,洗脱得率为72.63%(基于GPS-1质量计算),命名为GPS-1A。对GPS-1A进行降解,得到α-葡萄糖苷酶抑制率为91.58%的低分子质量多糖,降解得率为64.35%(基于GPS-1A质量计算),命名为DGPS-1A。
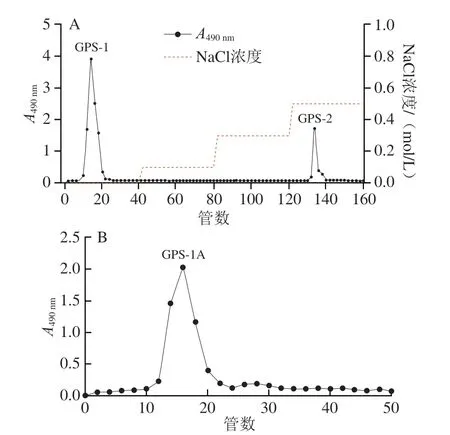
图1 GPS在DEAE-纤维素-52(A)和在Sephadex G-100(B)上的洗脱曲线Fig.1 Elution curves of GPS on DEAE-52 (A) and Sephadex G-100 column (B)
2.2 GPS的基本理化性质
由表1可知,GPS-1A和DGPS-1A的多糖质量分数接近,分别为94.84%和95.36%。然而,超声波辅助H2O2-VC处理后DGPS-1A中的糖醛酸的含量较GPS-1A显著提高了122.97%(P<0.05),这可能是H2O2-VC体系产生的羟自由基以及超声波的空化作用攻击糖苷键引起多糖暴露出更多的糖醛酸导致的。

表1 GPS、GPS-1A及DGPS-1A的得率及多糖、糖醛酸和蛋白质量分数Table 1 Yields and polysaccharide,uronic acid and protein contents of GPS,GPS-1A and DGPS-1A
2.3 GPS-1A和DGPS-1A的相对分子质量
研究发现,相对分子质量会影响多糖的生物活性和实际应用[23]。如图2所示,GPS-1A及其降解产物DGPS-1A的高效凝胶渗色谱图中均有一个对称峰,表明GPS-1A和DGPS-1A是均一的多糖组分。与GPS-1A相比,降解多糖DGPS-1A的色谱峰明显右移,相对分子质量分别为135(GPS-1A)和77.8(DGPS-1A)。结果表明,超声波辅助H2O2-VC反应可以降解GPS,并且降低其相对分子质量。超声波辅助H2O2-VC降解后,GPS相应的多分散度(重均分子质量(mw)/数均分子质量(mn))从3.19降低到2.00,这说明降解产物的分子质量分布逐渐变窄,变得更加均匀。

图2 GPS-1A和DGPS-1A的高效凝胶渗透色谱图Fig.2 High performance gel permeation chromatograms of GPS-1A and DGPS-1A
2.4 GPS-1A和DGPS-1A的单糖组成
研究表明,单糖作为多糖的天然基本单位,很大程度上影响着天然多糖的结构和功能活性[24]。如图3及表2所示,GPS-1A及其降解产物DGPS-1A的单糖组成相似,但物质的量比略有不同。GPS中单糖主要包括甘露糖(Man)、葡萄糖(Glc)、半乳糖(Gal)、阿拉伯糖(Ara)、岩藻糖(Fuc)、葡萄糖醛酸(GlcUA)和半乳糖醛酸(GalUA),其在GPS-1A及DGPS-1A中物质的量占比分别为0.15∶98.80∶0.36∶0.14∶0.09∶0.05∶0.41和1.35∶95.85∶0.54∶0.13∶0.73∶0.15∶1.17。单糖组成中葡萄糖的占比最高,证明GPS-1A和DGPS-1A是葡聚类中性多糖。降解前后单糖种类没有发生改变,表明超声波辅助H2O2-VC降解相对温和,几乎不破坏天然多糖的基本结构。降解后单糖物质的量占比发生了改变,DGPS-1A中葡萄糖占比下降,甘露糖、岩藻糖和半乳糖占比升高。这可能是因为羟自由基只攻击葡萄糖区附近的糖苷键,而其他中性多糖附近的糖苷键相对稳定,并未因羟自由基的存在而发生改变[13]。降解后葡萄糖醛酸和半乳糖醛酸占比升高,说明H2O2-VC产生的羟自由基作用于多糖主链葡萄糖区的糖苷键,释放出更多的糖醛酸,这与表1中糖醛酸质量分数变化的结果相印证。徐雅琴等[25]采用超声波辅助Fe2+-VC-H2O2降解黑加仑多糖,降解后多糖的单糖种类也未发生改变。
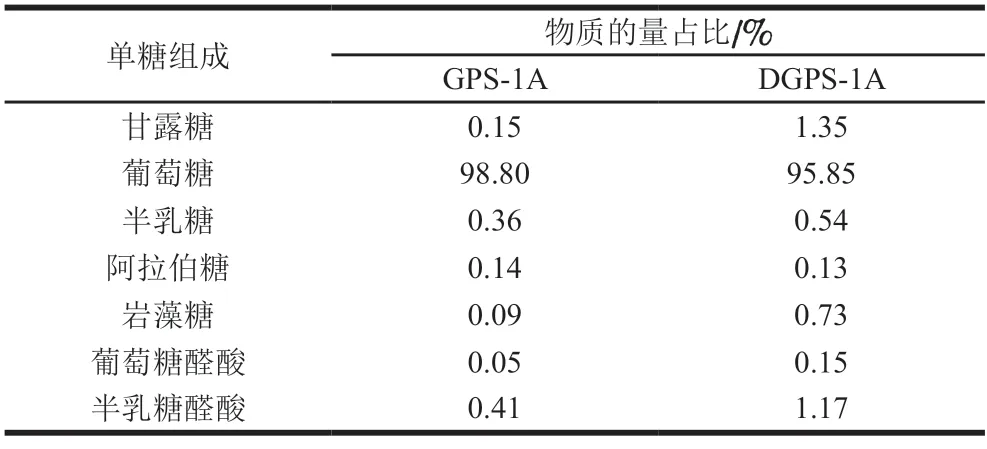
表2 GPS-1A和DGPS-1A的单糖组成Table 2 Monosaccharide compositions of GPS-1A and DGPS-1A
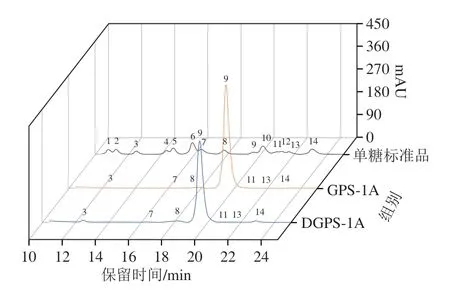
图3 GPS-1A和DGPS-1A单糖组成的高效液相色谱图Fig.3 High-performance liquid chromatographic profiles of monosaccharide compositions of GPS-1A and DGPS-1A
2.5 GPS-1A和DGPS-1A的傅里叶变换红外光谱分析
如图4所示,在4 000~500 cm-1范围内GPS-1A和D GPS-1 A 都呈现典型的多糖特征吸收峰[26]。在3 435.06 cm-1处有宽而强的吸收峰,为多糖O—H的伸缩振动峰;2 933.59 cm-1和1 591.2 cm-1处的吸收峰分别由C—H伸缩振动和C—O伸缩振动导致[27];1 350.11cm-1处的吸收峰为C—H的变角振动峰。GPS-1A在1 632.4 cm-1处的吸收峰相对较弱,说明其糖醛酸含量相对较少,与表1结果相印证。DGPS-1A在1 209.31 cm-1附近的吸收峰为双键=SO的对称伸缩振动,表明其可能存在硫酸基团[28]。在1 200~1 000 cm-1处的特征峰是由于C—O—C糖苷键和C—O—H侧基的伸缩振动导致的,表明GPS-1A和DGPS-1A为吡喃型多糖[29]。933.5 cm-1和829.3 cm-1处的特征吸收峰表明GPS-1A和DGPS-1A同时存在α-构型和β-构型的糖苷键[30]。综上,GPS-1A和DGPS-1A具有相似的特征峰,超声波辅助H2O2-VC处理不会影响GPS-1A的一级结构。
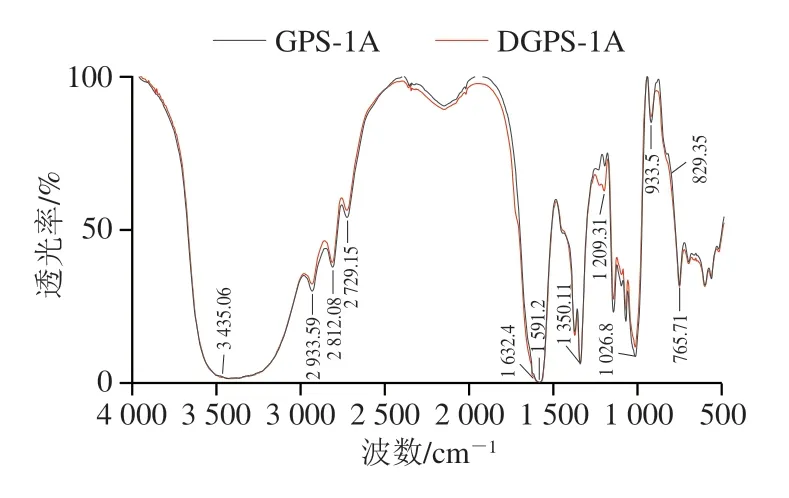
图4 GPS-1A和DGPS-1A的傅里叶变换红外光谱图Fig.4 Fourier transform infrared spectra of GPS-1A and DGPS-1A
2.6 GPS-1A和DGPS-1A的紫外光谱分析
如图5所示,纯化多糖GPS-1A和降解多糖DGPS-1A溶液在260、280 nm波长处未出现紫外吸收峰,说明多糖中不含核酸和蛋白质,这与2.2节GPS-1A和DGPS-1A中未检测出蛋白质的结果相印证。

图5 GPS-1A和DGPS-1A的紫外吸收光谱Fig.5 Ultraviolet absorption spectra of GPS-1A and DGPS-1A
2.7 GPS-1A和DGPS-1A的NMR氢谱分析
如图6所示,GPS-1A和DGPS-1A在δH3.5~5.3(图A、B)和δC60~110(图C、D)处的信号与多糖NMR信号的典型分布一致。通常情况下,化学位移δ大于5.0时表示多糖分子的异头氢为α-构型,而化学位移δ小于5.0则表示多糖分子的异头氢为β-构型[31]。由图6A、B可知,GPS-1A和DGPS-1A的1H-NMR图谱异头氢区域中存在4 个较为明显的信号峰,化学位移δ分别为3.59、3.77、3.90和5.33,说明GPS-1A和DGPS-1A中的糖苷键类型同时存在α-构型和β-构型,这与傅里叶变换红外光谱分析结果相印证。图6C、D结果表明GPS-1A由不同单糖组成,并且DGPS-1A检测出和GPS-1A相似的异头碳信号,结合1H-NMR图谱,得出DGPS-1A的异头氢所对应异头碳信号的化学位移主要为66.69、71.65、72.78和99.71,具体为δ99.71/5.33、δ72.78/3.59、δ71.65/3.77和δ66.69/3.90。根据先前的研究将其分别归属于α-D-GlcpA-(1→、→3,6)-β-D-Manp-(1→、→6)-α-D-Glcp-(1→和→3,5)-α-LAraf-(1→[32]。单糖组成中没有检出其他单糖类型,可能是由于其含量太低导致的。综上,超声波辅助H2O2-VC处理没有破坏GPS-1A的主要化学结构。
2.8 GPS-1A和DGPS-1A的表面形态
扫描电子显微镜可以定性分析多糖的表面形态。如图7所示,GPS-1A和DGPS-1A的表面形态非常相似,均具有光滑表面的片状和刚性棒状结构。但是,与GPS-1A(图7A~C)相比,DGPS-1A(图7D~F)中的碎片表面积明显增加,这可能是降解处理攻击多糖连接的糖苷键所导致的。以上结果表明,超声波辅助H2O2-VC降解改变了多糖的微观结构。

图7 GPS-1A(A~C)和DGPS-1A(D~F)的扫描电子显微镜图像Fig.7 Scanning electron microscopic images of GPS-1A (A-C) and DGPS-1A (D-F)
2.9 GPS-1A和DGPS-1A的刚果红和I2-KI分析
研究表明,多糖在溶液中可以表现出多种不同的构象,如单螺旋、双螺旋、三螺旋和聚集体等,其构象与多糖的生物活性有关[33]。通常,当三螺旋构象的多糖与刚果红结合时,稀碱溶液中的λmax红移,而浓碱溶液中的λmax显著降低。如图8A所示,GPS-1A+刚果红和DGPS-1A+刚果红的λmax均随着氢氧化钠浓度的升高而先升高再降低,最后趋于稳定,表明两种多糖中都存在三螺旋构象。且DGPS-1A的λmax低于GPS-1A,说明超声波辅助H2O2-VC处理后GPS-1A的三螺旋构象发生了一定的解体[34]。

图8 GPS-1A和DGPS-1A的结构表征Fig.8 Structure characterization of GPS-1A and DGPS-1A
I2-KI实验可以检测多糖中是否存在相对较长和多分支的结构。当分支更少、侧链更短的多糖与I2结合时,I2在350 nm波长处的吸收峰会移至565 nm波长处。如图8B所示,GPS-1A和DGPS-1A溶液在565 nm波长附近没有吸收峰,这表明两种多糖都具有复杂的结构,降解没有破坏GPS-1A的主要结构,两者主链上均有较多的分支和较长的侧链,而且没有淀粉。
2.10 GPS-1A和DGPS-1A的体外降血糖活性分析
糖尿病是一种慢性疾病,其特征是与饮食代谢异常相关的高血糖,并会导致慢性并发症的发生[35]。α-淀粉酶和α-葡萄糖苷酶抑制剂会抑制小肠中碳水化合物水解酶的活性,从而减少淀粉中碳水化合物的释放,并延缓碳水化合物的吸收[36]。因此,天然的α-淀粉酶和α-糖苷酶抑制剂引起了人们的极大关注。
如图9所示,所有样品对α-淀粉酶和α-葡萄糖苷酶均表现出剂量依赖的抑制作用,且DGPS-1A对α-淀粉酶和α-葡萄糖苷酶的抑制活性显著高于GPS-1A(P<0.05)。当质量浓度为6.0 mg/mL时,GPS-1A、DGPS-1A和阿卡波糖对α-淀粉酶的抑制率分别为(54.04±1.97)%、(78.03±2.29)%和(90.27±1.94)%,半抑制浓度(half maximal inhibitory concentration,IC50)分别为4.34、1.98、0.04 mg/mL。在最高质量浓度(6 mg/mL)下,GPS-1A、DGPS-1A和阿卡波糖对α-葡萄糖苷酶的抑制率最高,分别为(65.16±2.53)%、(90.83±3.04)%和(97.26±1.21)%,相应的IC50值分别为1.44、0.51、0.07 mg/mL。
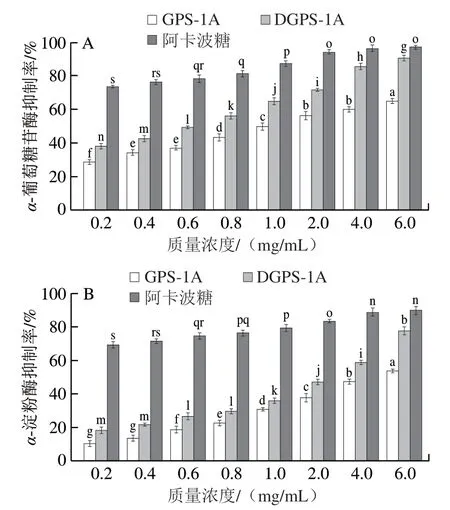
图9 GPS-1A、DGPS-1A对α-葡萄糖苷酶(A)和α-淀粉酶(B)活性的抑制作用Fig.9 Inhibitory effects of GPS-1A and DGPS-1A on the activities of α-glucosidase (A) and α-amylase (B)
多糖可以与α-淀粉酶或α-葡萄糖苷酶结合改变其酶结构,也可以与底物和酶结合形成三元络合物,导致酶催化能力下降[37]。降解后的多糖相对分子质量减小、表面积增大,可以结合更多的酶分子,阻碍其和底物接触反应,从而提高酶的抑制率。毛美林等[35]采用H2O2-VC法制备江蓠降解多糖,发现中等分子质量的降解多糖GLP1具有最强的α-葡萄糖苷酶抑制作用。以上结果说明,多糖的降血糖活性不仅与分子质量有密切关系,还与不同来源多糖特定的结构有关。
2.11 GPS-1A和DGPS-1A的体外抗氧化活性分析
前人研究发现GPS具有一定的抗氧化能力[38]。因此,本研究并比较了超声波辅助H2O2-VC处理对GPS-1A抗氧化活性的影响。如图10所示,GPS-1A和DGPS-1A的DPPH自由基清除率、羟自由基清除率和还原能力均呈剂量依赖关系,并且降解后的DGPS-1A抗氧化活性更好,这说明超声波辅助H2O2-VC处理明显提高了多糖的抗氧化能力,与文献[17]的结果相似。

图10 GPS-1A和DGPS-1A的体外抗氧化活性分析Fig.10 In vitro antioxidant activity of GPS-1A and DGPS-1A
DPPH自由基作为一种稳定的自由基,被广泛用于测定各种抗氧化剂清除自由基的能力[39]。如图10A所示,GPS-1A、DGPS-1A和阳性对照(VC)对DPPH自由基表现出较强的清除能力,当质量浓度达到6.0 mg/mL时三者的DPPH自由基清除率分别为(31.77±2.42)%、(89.56±1.17)%和(99.50±0.66)%,其IC50值分别为12.45、2.32、0.03 mg/mL。Xu Yaqin等[40]对黑加仑多糖的研究也表明,相对分子质量较低的黑加仑多糖DBCP-2具有更强的DPPH自由基清除能力,本研究结果与之一致。
在活性氧化物中,羟自由基具有最高的氧化活性,它可以非特异性地攻击邻近的生物分子,并对细胞造成氧化损伤[41]。如图10B所示,在0.2~6.0 mg/mL的质量浓度下,所有样品均对羟自由基有不同程度的清除能力,其清除能力排序依次为GPS-1A<DGPS-1A<VC,其IC50值分别为15.84、1.69、0.05 mg/mL。在质量浓度为6.0 mg/mL时,GPS-1A、DGPS-1A和VC的羟自由基清除率分别为(30.85±2.29)%、(76.85±1.63)%和(99.36±0.90)%。在多糖的官能团中,还原羟基可能通过提供电子将自由基还原为更稳定的形式,或通过直接与自由基反应以终止自由基链反应来参与抗氧化作用[42]。本研究结果表明,降解后的多糖DGPS-1A具有更多的还原羟基和更大的比表面积,所以具有更强的羟自由基清除能力。
化合物的还原能力可作为抗氧化活性的重要指标[41]。反应液的吸光度越高,表明反应化合物的还原能力越强。如图10C所示,当质量浓度为6.0 mg/mL时,DGPS-1A的还原能力比GPS-1A提高了1.03 倍,说明相对分子质量降低可使还原能力增强。多糖链通常有一个还原末端和一个非还原末端,低分子质量的多糖由于具有更多的还原和非还原末端而显示出更强的还原能力[43]。
3 结论
本研究以人参为原料得到粗多糖GPS,通过分离纯化得到了中性组分GPS-1A,并采用超声波辅助H2O2-VC法在特定降解条件下降解得到DGPS-1A。GPS-1A和DGPS-1A的相对分子质量分别为135和77.8。两种多糖的单糖种类一致,但物质的量比略有不同。结构分析表明降解体系中生成的羟基虽然没有改变多糖的主要官能团结构,但攻击了主链糖苷键,使多糖的相对分子质量减小、表面积增大,从而提高了DGPS-1A的体外降血糖和抗氧化活性(与GPS-1A相比)。综上,DGPS-1A具有开发为新型抗氧化剂和抗糖尿病剂的潜在价值。
猜你喜欢
汽车电器(2019年1期)2019-03-21
天然产物研究与开发(2019年1期)2019-03-01
中成药(2018年5期)2018-06-06
中成药(2017年8期)2017-11-22
光谱学与光谱分析(2016年5期)2016-07-12
天然产物研究与开发(2016年1期)2016-06-05
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
河南科技(2015年8期)2015-03-11
天然产物研究与开发(2014年3期)2014-04-27
茶叶通讯(2014年4期)2014-02-27
