
ALK5小分子抑制剂生物活性研究进展
2023-11-03 04:21李倩倩薛晓文中国药科大学药学院南京200中国药科大学药物化学教研室南京200
中南药学 2023年10期
李倩倩,薛晓文,2*(.中国药科大学药学院,南京 200;2.中国药科大学药物化学教研室,南京 200)
转化生长因子-β(transforming growth factor-β,TGF-β)是一种多功能细胞因子,广泛存在于哺乳动物的组织细胞和转化细胞中,自20世纪70年代末被发现以来,已在哺乳动物中发现了33个该家族成员配体,包括TGF-β、骨形态发生蛋白(bone morphogenetic protein,BMP)、激活素、抗缪勒式管激素(anti-Müllerian hormone,AMH)、生长分化因子(growth and differentiation factors,GDF)和结节素(nodal)等。这些配体具有相似的结构,并共享信号转导的基本机制,在生物体生殖细胞、胚胎和各种组织或器官中广泛表达,与细胞增殖、分化、转分化、细胞外基质(extracellular matrix,ECM)重塑、衰老和细胞凋亡等多种细胞行为密切相关[1]。TGF-β配体通过与细胞膜表面的特异性受体(即TGF-β受体)结合才能发挥生理效应。ALK5为TGF-βⅠ型受体中研究最为广泛的一个亚型,抑制该亚型可以产生抗肿瘤等作用。本文在介绍TGF-β、相应受体及相关信号通路的基础上,对近年来报道的ALK5抑制剂的各种生物活性进行梳理,希望为今后的ALK5抑制剂研究提供有益参考。
1 TGF-β、TGF-β受体及相关信号通路
TGF-β是一种由两个含有112个氨基酸的单体通过二硫键连接的蛋白质,目前已发现6种亚型,其中TGF-β1(占90%以上,活性最强)、TGF-β2 和TGF-β3 3种亚型常见于哺乳动物体内,它们能够通过与TGF-βⅠ型受体(TβRⅠ,也称为作为激活素受体样激酶,ALK)和Ⅱ型受体(TβRⅢ)这两种类型的跨膜受体的结合来触发细胞内信号[2-3]。目前,已经鉴定出7个TβRⅠ亚型(ALK1~ALK7)和5个TβRⅡ亚型,这两种受体在所有细胞中均能表达,它们可以与相应的TGF-β超家族成员形成特定的异性复合物进行信号传递,从而发挥相应的细胞功能。初始合成的TGF-β是以非活性前体的形式存在(即潜伏期TGF-β),需要通过潜伏TGF-β结合蛋白(latent TGF-βbinding protein,LTBP)和潜伏期相关肽(latency-associated peptide,LAP)组成的复合物激活后才能发挥相应的生物效应[4]。激活后的TGF-β首先与TβR Ⅱ结合进行信号传导,将ALK5募集到高度保守的富含Gly-Ser的近膜区域(GS区)使其发生构象调节并激活[5]。然后,被激活的ALK5将信号进一步传递给其下游底物SMAD蛋白——SMAD2和SMAD3,便完成了经典的SMAD信号通路传导(见图1)[6]。
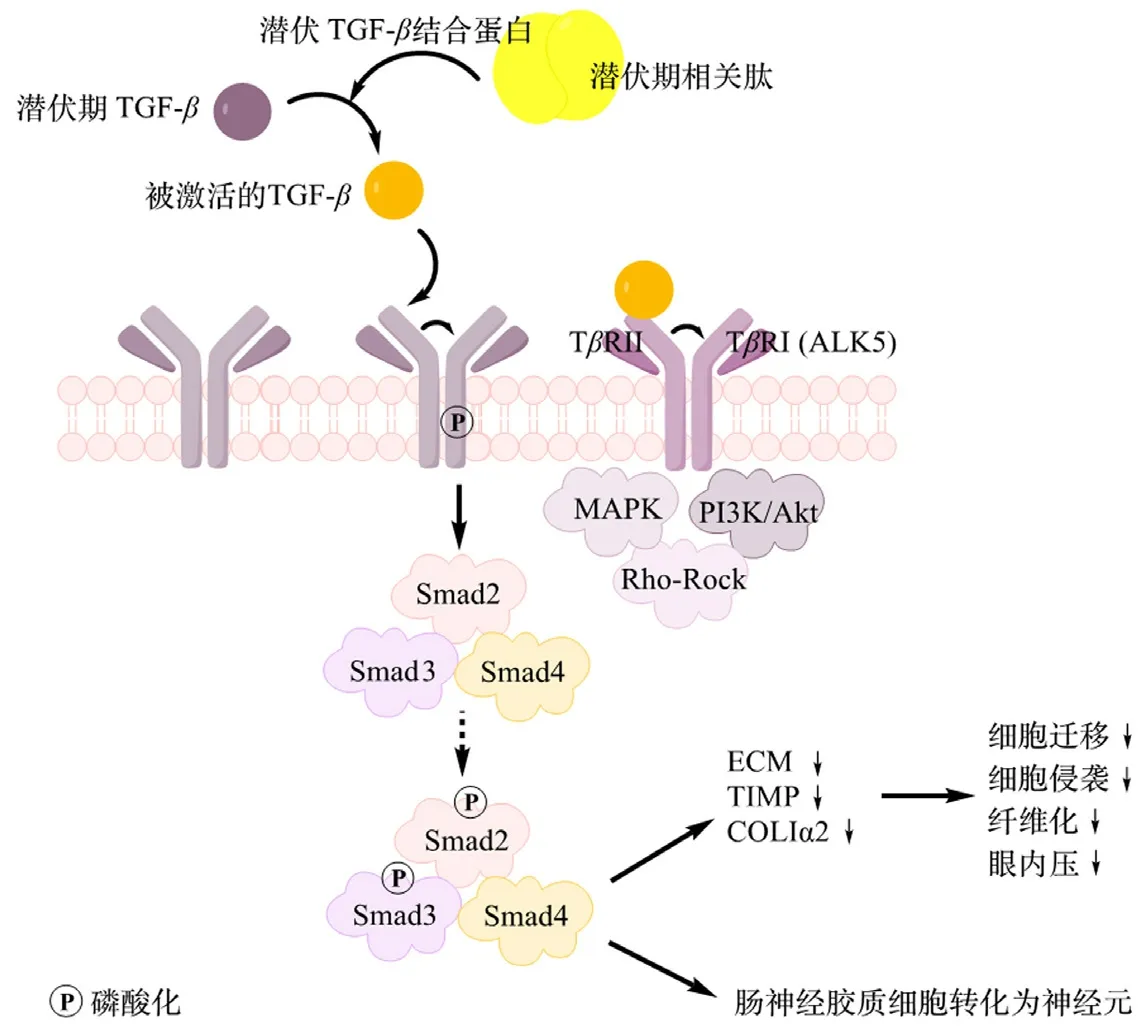
图1 TGF-β/Smad 信号通路Fig 1 TGF-β/Smad signaling pathway
除了经典的SMAD信号通路,TGF-β还可以通过丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)、磷脂酰肌醇-3-激酶/蛋白激酶B(phosphatidylinositol 3 kinase/protein kinase B,PI3K/Akt)、Rho-Rock等激活非SMAD途径,从而影响TGF-β因子下游的一些关键细胞的生长发育过程[7]。由于ALK5在TGF-β信号通路中起着至关重要的作用,因此被视为治疗多种癌症、纤维化疾病的重要靶点。近年来,ALK5抑制剂在治疗其他疾病如青光眼、血脑屏障损伤及肠道疾病等方面也取得了一定的进展[8]。
2 ALK5抑制剂的生物活性
2.1 抗肿瘤作用
TGF-β是一种众所周知的肿瘤促进因子,如何通过抑制其活性而发挥抗肿瘤作用一直都是研究热点。体外研究显示,ALK5抑制剂能够有效阻断SMAD蛋白的磷酸化,从而减少肿瘤的转移,抑制肿瘤细胞的增殖。此外,ALK5抑制剂还可以通过阻断癌细胞的免疫逃逸来防止肿瘤侵袭[9]。到目前为止,进行临床试验的ALK5小分子抑制剂有化合物Vactosertib(EW-7197,1)、Galunisertib(LY-2157299,2)、LY-3200882(3)和PF-06952229(4)(见图2),且临床试验均为实体瘤方向。其中,LY-3200882因公司战略调整,于2019年停止产品开发(NCT04158700)。另一产品PF-06952229早先用于治疗具有高TGF-β特征和上皮-间充质转化(EMT)表达的晚期或转移性癌症患者(NCT03685591),作为单一疗法并与其他治疗药物联合使用,后也因公司的战略调整,于2021年停止了该产品的开发。

图2 ALK5小分子抑制剂Fig 2 ALK5 small molecule inhibitors
Vactosertib是一种口服的可逆性TGF-βⅠ型受体酪氨酸激酶抑制剂,IC50为13 nmol·L-1[10],用于治疗晚期/转移性癌症等。目前,针对晚期/转移性非小细胞肺癌(NSCLC)、转移性胰腺导管腺癌和骨髓增生异常综合征等多种实体瘤的临床试验正在展开。2017年,该化合物在美国被授予孤儿药称号,用于治疗肝细胞癌。2020年,Vactosertib/紫杉醇联合用药在美国获得孤儿药资质,用于治疗转移性胃腺癌。2021年,该产品被指定为孤儿药,用于治疗胰腺癌。最近,Park等[11]研究表明,口服Vactosertib可以通过抑制氧化应激、EMT、癌症干细胞(CSC)和纤维化来克服放疗在乳腺癌治疗中的局限性,因此,Vactosertib联合放疗在临床上可能更适用于乳腺癌患者。
目前礼来公司对于Galunisertib(IC50=56 nmol·L-1)的研究正处于Ⅱ期临床开发阶段(NCT01246986),用于治疗肝细胞癌、胶质瘤和多形性胶质母细胞瘤,而该药物用于骨髓增生异常综合征治疗的Ⅱ/Ⅲ期临床试验已经完成[12]。用于治疗转移性胰腺癌、神经胶质瘤、结直肠癌、乳腺癌、转移性去势抵抗性前列腺癌和子宫或卵巢癌肉瘤等的早期临床试验也已经或正在进行中。2013年,作为欧盟指定孤儿药,Galunisertib用于治疗肝细胞癌和神经胶质瘤。
目前处于临床前研究的ALK5抑制剂较多,其中有3个结构值得关注。Zhang等[13]利用基于分子对接的虚拟筛选对商业天然产物库进行筛选,发现黄酮苷山柰酚3-O-龙胆双糖苷(KPF 3-O-G,5)(见图2,IC50=2.589 mol·L-1)是一种有效的ALK5抑制剂,这是近年来发现的为数不多的天然产物类ALK5抑制剂。该化合物能够通过直接与ALK5的ATP位点结合,抑制Smad2的磷酸化以及Smad4的表达,从而减少细胞增殖并抑制TGF-β诱导的细胞迁移和侵袭。体内研究表明,KPF 3-O-G给药(40 mg·kg-1)能够减少人卵巢癌异种移植小鼠模型中的肿瘤生长,且无明显毒副作用,表明KPF3-O-G具有开发成为肿瘤治疗药物的潜力。Xu等[14]以LY-3200882为先导,通过对其进行药效团分析和分子对接研究,对分子中的连接环进行替换,得到了一系列4-(吡啶-4-氧基)-3-(3,3-二氟环丁基)吡唑衍生物,其中活性最好的化合物6(见图2)(IC50=30 nmol·L-1)的体外、体内活性及药代动力学特征均优于LY-3200882,表现出良好的抗肿瘤疗效。此外,由于p38α丝裂原活化蛋白激酶(p38αMAPK)对肿瘤坏死因子-α具有重要调控作用,因此,抑制p38αMAPK被视为治疗肿瘤的一种有效手段[15-16]。近年来,对于ALK5及p38αMAPK双靶点抑制剂的研究,也取得了较好的结果。Liu等[17]在前人研究的基础上,设计合成了4-(1H-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1H-咪唑和4-(1-甲基-1H-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1H-咪唑两个系列的化合物,并评估了它们对ALK5和p38αMAPK的抑制作用,结果显示,化合物J-1090(7,见图2)对两者均表现出了强大的抑制作用,IC50值均为4 nmol·L-1。进一步的体外研究表明,该化合物能够通过抑制TGF-β诱导的EMT来抑制肿瘤细胞的迁移和侵袭。
2.2 抗纤维化作用
TGF-β1最初被发现时,便因其能够刺激软琼脂中培养的大鼠成纤维细胞的生长,而被认为是纤维化的主要细胞因子[18]。研究表明,在多种ECM积累引起纤维化的器官中都能够观察到TGF-β的过表达[19-20]。因此,抑制ALK5不失为对抗各种器官纤维化的有效策略。目前,抗纤维化作用的ALK5小分子抑制剂均处于临床前研究阶段,尚未有药物进入临床研究阶段。
Yue等[21]在MPLW515L和JAK2V617F小鼠模型中,发现Galunisertib无论在体内还是体外,均能够消除由于骨髓纤维化引起的胶原蛋白Ⅰ和胶原蛋白Ⅲ的持续过量产生,明显改善了小鼠的骨髓纤维化程度,为阻断ALK5作为骨髓纤维化治疗策略提供了临床前依据。Ferreira等[22]使用GW-788388(8,见图2)对克氏锥虫(来自哥伦比亚毒株的102种寄生虫)感染后的小鼠进行口服治疗(3 mg·kg-1),发现GW-788388 有效逆转了富含连接蛋白-43的细胞间斑块的损伤,并减少了心脏组织的纤维化,因此,抑制TGF-β通路可能是美洲锥虫病心脏病慢性期心脏纤维化治疗的重要且可靠的策略。
Rezníckova等[23]设计并合成了一系列以2,3,4-取代的5,5-二甲基-5,6-二氢-4H-吡咯并[1,2-b]吡唑结构为核心的衍生物,其中化合物9(见图2)活性最好,对ALK5的IC50值为80 nmol·L-1。细胞研究表明,这些化合物能够通过引起Smad2/3的剂量依赖性去磷酸化,阻断Smad2/3向TGF-β刺激的细胞核的转位,呈现出抗纤维化作用。Zhu等[24]报道了一系列含有[1,2,4]三唑并[1,5-a]吡啶-6-基部分的化合物,并评估了这些化合物对ALK5和p38α激酶的抑制活性,发现在吡唑环的4-位插入苯并[c][1,2,5]噻二唑-5-基部分可显著提高ALK5激酶的活性。其中最有效的化合物10(见图2)的IC50值为30 nmol·L-1,对p38α激酶的选择性指数为235,是LY-2157299的59倍,与EW-7197相当。免疫细胞化学染色、蛋白质印迹和RT-PCR测定结果均显示化合物10有效抑制了TGF-β诱导的肝星状细胞(HSC)活化,对肝纤维化治疗表现出极高的治疗潜力,有望成为治疗肝纤维化的临床前候选药物。Zheng等[25]设计合成了多种噻吩并[3,2-c]吡啶-2-基吡唑衍生物,并评价了它们对ALK5的抑制活性,发现化合物 J-1063(11,见图2)(IC50=30 nmol·L-1)能够有效抑制TGF-β诱导的活化HSC的纤维化和炎症,减少硫代乙酰胺诱导的小鼠肝纤维化中巨噬细胞和中性粒细胞的浸润,其延缓肝纤维化的进展及抑制伴随的炎症的能力明显优于Galunisertib。该化合物不仅具有优越的药理作用,而且合成相对简单,是治疗和预防肝纤维化的有希望的候选药物。
Xie等[26]通过虚拟筛选和药效团构建设计并合成了一种新型ALK5小分子抑制剂Cpd-0225(12,见图2),生物活性研究表明该化合物能够通过阻断Smad依赖性和非Smad依赖性信号通路,发挥肾保护作用,在体外或体内均能发挥作用,且在相同剂量下,比SB-431542(13,见图2)具有更好的抗纤维化作用,有望成为治疗慢性肾病的有效临床候选药物。此外,由于对成纤维细胞具有较好的抑制作用,SB-431542最新用于青光眼小梁切除术后滤过泡瘢痕化的防治,且有望替代早先使用的丝裂霉素C等细胞毒类药物,因为ALK5抑制剂具有更低的细胞毒性[27]。
2.3 促进肠神经胶质细胞转化为神经元
肠神经系统(enteric nervous system,ENS)由肠壁中的肌间神经丛和黏膜下神经丛组成,该系统主要包含肠胶质细胞和肠神经元。多种不良因素,如炎症、衰老、饮食改变、代谢紊乱等,都可能导致肠神经元损伤或ENS功能失调,从而引起胃肠动力和感觉功能障碍,如胃轻瘫、肠易激综合征和先天性巨结肠等[28-30]。对于患有先天性巨结肠的儿童,手术切除整个肠道结肠或部分结肠仍然是治疗的主要选择,但这种治疗不能预防生命后期发生的小肠结肠炎、便秘或失禁等终生并发症。尽管在成人肠道中产生新神经元的手段相对有限,但添加新的肠神经元来替代受损的神经元将仍是修复ENS功能障碍的解决方案。
Medina等[31]研究发现,SD-208(14,见图2)能够通过TGF-β/ALK5/SMAD通路参与实验性肠纤维化的发病机制,在厌氧菌和三硝基苯磺酸诱导的结肠炎的动物模型中,有效抑制由于Smad2和Smad3的磷酸化以及ALK5蛋白、组织金属蛋白酶抑制剂-1(tissue inhibitor of matrix metalloproteinase,TIMP)和Ⅰ型胶原α2(α2 type 1 collagen,COL1α2)重组蛋白表达上调产生的纤维化。Shi等[32]发现在正常成年小鼠模型中,用RepSox(15,见图2)3及10 mg/(kg·d)灌胃,持续给药2周,结果显著促进了ENS中肠神经胶质细胞向神经元的转化,并对小鼠的胃肠道运动产生了一定的影响。EW-7197和LY-257299也能产生与SD-208相似的效果,并且利用shRNA下调肠神经胶质细胞中ALK5的表达也会产生类似效果,进一步证实了ALK5信号通路在肠神经胶质细胞向神经元的转变中发挥了重要作用。这些结果表明,抑制ALK5可能是一种有前途的治疗肠道疾病的有效策略。
2.4 血脑屏障保护作用
研究表明,星形胶质细胞中TGF-β受体的条件性基因敲除或TGF-β信号传导的药理学抑制均能够逆转老年小鼠的血脑屏障受损(blood brain barrier disruption,BBBD)。同时,在接受TGF-β抑制治疗的患有血脑屏障(blood brain barrier,BBB)的老年人群受试者中,发现同样的信号通路被激活[33]。
Atis等[34]通过使用SB-431542对血管紧张素(angiotensin,ANG)Ⅱ诱导的高血压小鼠进行治疗,发现SB-431542能够降低由于ANGⅡ输注引起的BBB对白蛋白Alexa fluor 594的渗透性的增加,因此抑制TβRⅠ可以防止高张力条件下BBBD,可作为一种新的治疗方式来保护高血压患者的大脑。Preininger等[35]使大鼠海马体长期暴露于血清白蛋白,通过TGF-β信号传导诱导星形胶质细胞应激诱导的早衰,发现在对BBBD大鼠模型每日使用ALK5抑制剂IPW-53715(结构未见公布)进行给药后,海马组织中衰老星形胶质细胞的负担显著减轻,这也进一步验证了ALK5抑制剂IPW-5371能够有效预防BBBD。
2.5 降低眼压作用
青光眼是一种进行性眼部疾病,可导致不可逆的视网膜神经节细胞死亡,是全球视力丧失的主要原因。由于现有药物或治疗方法均会产生耐药性或者不同程度的不良反应如心律失常和支气管收缩等,因此亟需开发治疗青光眼的新疗法[36-37]。
眼内压(intra-ocular pressure,IOP)升高是青光眼发生和发展最重要的危险因素,降低IOP已被证明可有效治疗青光眼。IOP由房水的产生和流出控制,小梁网是房水从眼睛排出的主要途径。Aoshima等[38]研究表明,ALK5抑制剂SB-431542能够通过抑制小梁网中ECM的产生达到降低IOP的目的,并且其作用优于Rho相关的卷曲蛋白激酶抑制剂Y-27632,每日一次重复使用可增强小鼠的IOP降低效果。因此,ALK5抑制剂或许可以作为治疗青光眼的新疗法进行开发。
3 小结与展望
TGF-β几乎渗透到人体内各生物学过程中的方方面面,形成了一个巨大的信号网络,其生物活性的研究一直都在进行中。ALK5作为TGF-β/Smad信号通路中不可或缺的一环,其抑制剂在TGF-β抑制剂中占据着举足轻重的地位。虽然至今尚未有ALK5小分子抑制剂被批准上市,但目前在研药物中,EW-7197和LY-2157299在临床试验中均有良好的表现,并且不少活性好的新型小分子抑制剂也不断被发现。尤其是具有ALK5抑制活性的天然产物也逐渐被发现,对其进行结构改造也呈现出广阔的前景。在生物活性方面,ALK5小分子抑制剂除了具有抗肿瘤和纤维化作用以外,在肠道疾病、BBBD和青光眼治疗等方面也有一定的价值。总之,随着对ALK5小分子抑制剂的研究深入,更多安全有效的ALK5小分子抑制剂将进入临床发挥治疗作用。
猜你喜欢
传染病信息(2022年3期)2022-07-15
中学生数理化·中考版(2021年12期)2021-12-31
中学生数理化·中考版(2021年11期)2021-12-06
肝博士(2021年1期)2021-03-29
中学化学(2017年6期)2017-10-16
上海农业学报(2017年3期)2017-04-10
医学研究杂志(2015年6期)2015-07-01
中国当代医药(2015年16期)2015-03-01
中国药理学通报(2014年2期)2014-05-09
中国中医药现代远程教育(2014年13期)2014-03-01
