
BTK抑制剂LOXO-305的合成工艺研究
2023-11-02 02:53:32项良华姜虹羽唐春雷
化工时刊 2023年4期
项良华 王 霞 袁 昕 姜虹羽 王 杰 唐春雷
(江南大学 生命科学与健康工程学院,江苏 无锡 214122)
LOXO-305(1)的化学结构如图1所示,是由Redx Pharma公司开发,后被礼来公司收购的一种处于3期临床研究阶段的口服非共价布鲁顿酪氨酸激酶(BTK)抑制剂。BTK是一种非受体蛋白酪氨酸激酶(Tec)家族的细胞质激酶,由调节不同信号通路(包括PI3K、MAPK和NF-κB等)的造血细胞和浆细胞表达[1, 2]。BTK是B淋巴细胞(B细胞)受体通路的重要信号分子,在B细胞的各个发育阶段表达,B细胞的异常激活被证明在B细胞恶性肿瘤、自身免疫性疾病和炎症的发病机制中起核心作用[3]。过去十年已经开发了相当数量的BTK抑制剂,第一代和第二代主要是共价抑制剂(图2),第三代主要是非共价抑制剂,其开发处于早期阶段。伊布替尼(2)是首创的共价BTK抑制剂,已被批准用于治疗套细胞淋巴瘤和边缘区淋巴瘤,目前正在研究用于其他血液系统恶性肿瘤患者[4]。然而,伊布替尼的治疗相关不良反应(AE)限制了其在癌症治疗中的应用,在临床研究中,一些不良事件导致大量(9~23%)患者停止治疗[5-8]。
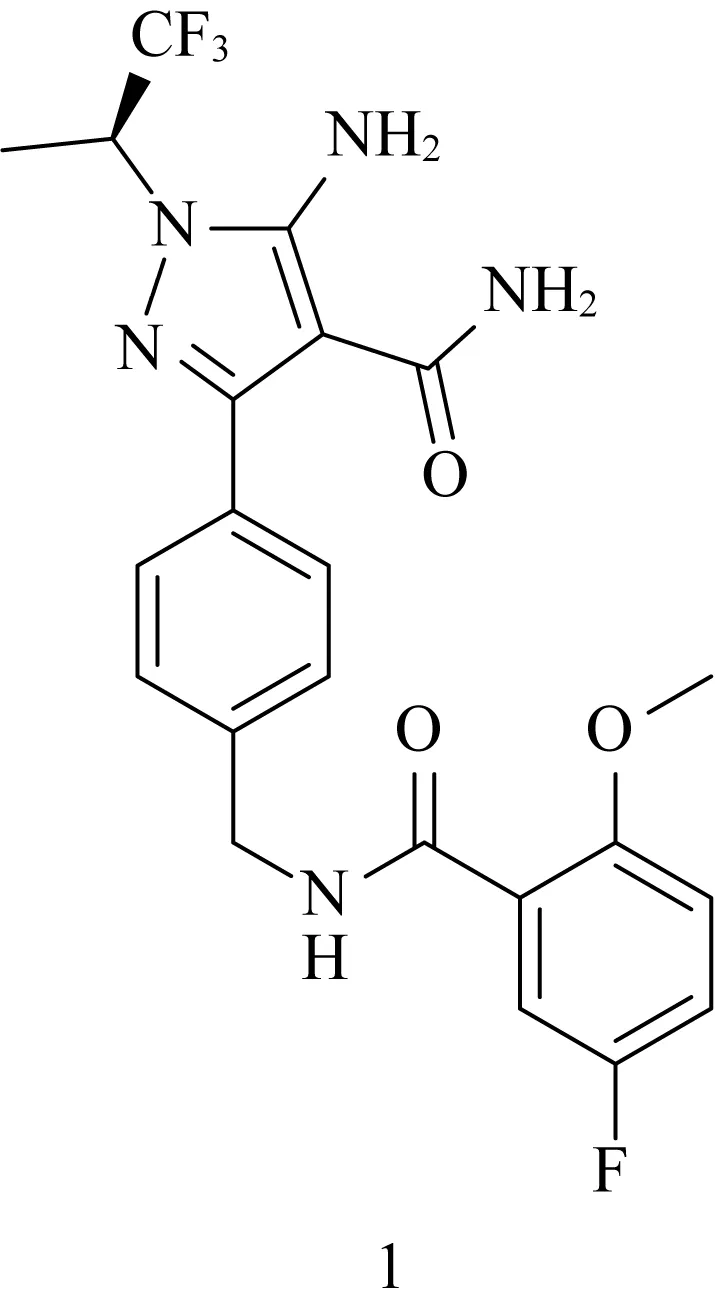
图1 LOXO-305Fig. 1 LOXO-305
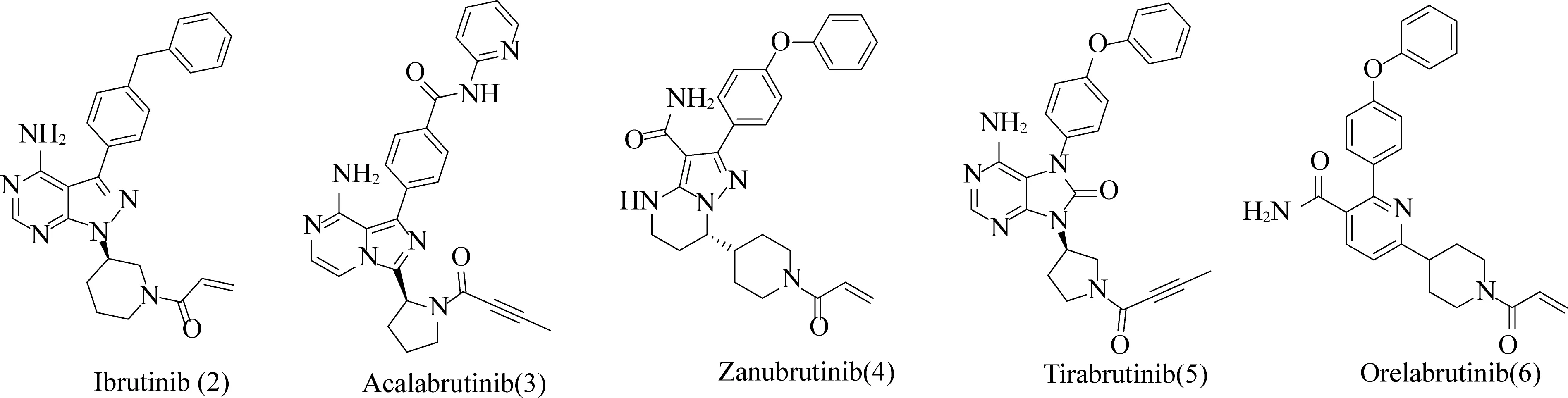
图2 目前上市的BTK抑制剂Fig. 2 BTK inhibitors currently on the market
此外,耐药突变体的产生减少了它们的使用,特别是,用丝氨酸残基等位替换481号位半胱氨酸(Cys481)降低了不可逆BTK抑制剂的主要活性。然而,不与Cys481相互作用的可逆抑制剂可以抑制C481R、T474I和T474M突变体,这成为一种新的开发方向。LOXO-305,一种可逆抑制剂,适用于BTK和BTK的不同变体C481突变,具有高靶向效力(IC50<1×10-9mol·L-1),对体外检测的370种激酶中超过98%表现出大于300倍的选择性,在1 mmol·L-1时对非激酶脱靶没有显著抑制[9]。LOXO-305有望在对伊布替尼产生耐药性而复发的疾病中表现出有效的治疗活性。本文对LOXO-305的合成方法进行研究,以期为该化合物及结构类似物的研究提供理论参考。
1 合成路线
目前,只有原研公司报道了LOXO-305的合成路线[10](图3):以5-氟-2-甲氧基苯甲酸(X1)为起始原料,在氮气氛围下以乙腈(ACN)为溶剂,与二氯亚砜(SOCl2)反应,得到酰氯衍生物X2,然后4-氨甲基苯甲酸与X2偶联得到产物X3;产物X3再以同样的条件制成X4,随后与丙二腈偶联得到中间体X5;中间体X5在92 ℃条件下与原甲酸三甲酯(TMOF)反应得到产物X6;产物X6与2,2,2-三氟-1-甲基乙基肼在三乙胺(TEA)的催化下环合得到环合产物X7;环合产物X7再经氰基水解得到终产物LOXO-305。
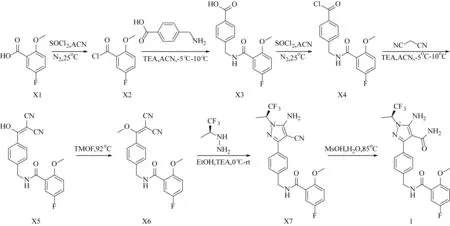
图3 LOXO-305的合成路线Fig. 3 Synthetic route of LOXO-305
原研专利的合成路线步骤为七步反应,较为烦琐,增加了工艺成本,另外部分中间体需要经过两次酰氯反应,容易产生大量毒废气体,不利于环保,限制了该合成路线在工业生产中的应用。作者同样以5-氟-2-甲氧基苯甲酸(X1)为起始原料,经两步缩合反应即可得到关键中间体N-((4-(2,2-二氰基-1-羟基乙烯基)苯基)甲基)-5-氟-2-甲氧基苯甲酰胺(X5)(图4),反应条件温和,避免了使用酰化试剂,且收率高达90.4%,大大降低了工艺成本。另外,4-氨甲基苯甲酸原料在有机溶剂中的溶解度极差,大大延长了反应时间,影响收率。改进工艺中选择氢氧化钠(NaOH)水溶液作溶剂,能很好地溶解该原料,在室温下仅3 h就能反应完全。优化之后的工艺路线不仅提高了总收率,降低了生产成本和能耗,而且反应条件相对温和,后处理操作简便,适合工业化制备。

图4 LOXO-305优化的合成路线Fig. 4 Optimized synthetic route of LOXO-305
2 合成实验
2.1 主要仪器与试剂
LCMS-80质谱仪,日本岛津公司;LC1260色谱仪,美国安捷伦科技有限公司;Bruker AVII-400 MHZ核磁共振仪,德国Bruker公司,TMS为内标。实验所用试剂均为市售分析纯或化学纯,未经进一步纯化处理。
2.2 合成步骤
2.2.1 4-(((5-氟-2-甲氧基苯甲酰基)氨基)甲基)苯甲酸(X3)的制备
5-氟-2-甲氧基苯甲酸(5g,29 mmol)溶于60 mL四氢呋喃(THF)中,冰浴条件下加入N,N-羰基二咪唑(CDI)(4.7 g,29 mmol)搅拌30 min,将上述反应液缓慢滴加至4-氨甲基苯甲酸(4.4 g,29 mmol)的NaOH溶液中(1 mol·L-1),滴毕,移至室温反应3 h。薄层色谱法(TLC)监测原料反应完毕。用5 mol·L-1HCl调pH至3~4,溶液变澄清,减压浓缩至体积20 mL左右,大量固体析出,抽滤,滤饼用乙醇(EtOH)淋洗2次,置于真空干燥箱干燥过夜,得白色固体8.7 g,收率98.2%。ESI-MS m/z: 304.1[M+H]+。1H NMR (400 MHz, DMSO-d6):δ12.85 (s, 1H), 8.87 (t,J=6.1 Hz, 1H), 7.91 (d,J=8.1 Hz, 2H), 7.50 (dd,J=9.2, 3.3 Hz, 1H), 7.44 (d,J=8.0 Hz, 2H), 7.37~7.31 (m, 1H), 7.19 (dd,J=9.1, 4.3 Hz, 1H), 4.56 (d,J=6.1 Hz, 2H), 3.89 (s, 3H).
2.2.2 N-((4-(2,2-二氰基-1-羟基乙烯基)苯基)甲基)-5-氟-2-甲氧基苯甲酰胺(X5)的制备
在氮气氛围,冰浴条件下向1-羟基苯并三唑(HOBT)(3.8 g,29 mmol)、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDCI)(5.3 g,29 mmol)、X3(8.5 g,28 mmol)和丙二腈(2.2 g,34.8 mmol)的THF(100 mL)溶液中缓慢滴加TEA(8.5 g,87 mmol),滴毕,将混合物移至室温下搅拌反应6 h。TLC监测原料反应完毕。减压浓缩,加入乙酸乙酯溶解,用饱和碳酸氢钠(NaHCO3)水溶液洗一次,饱和氯化铵(NH4Cl)水溶液洗一次,有机相再用5 mol·L-1HCl洗涤一次,有机相用无水硫酸钠干燥,抽滤,减压浓缩得白色固体9.1 g,收率92.1%。ESI-MS m/z: 352.1 [M+H]+。1H NMR (400 MHz, DMSO-d6):δ8.80 (t,J=6.2 Hz, 1H), 7.56~7.46 (m, 3H), 7.37~7.26 (m, 3H), 7.17 (dd,J=9.1, 4.3 Hz, 1H), 4.50 (d,J=6.1 Hz, 2H), 3.88 (s, 3H).
2.2.3 N-((4-(2,2-二氰基-1-甲氧基乙烯基)苯基)甲基)-5-氟-2-甲氧基苯甲酰胺(X6)的制备
将X5(9 g,25.6 mmol)加入原甲酸三甲酯(TMOF)(80 mL)的溶液中,升温到100 ℃搅拌反应10 h。TLC监测原料反应完毕。减压浓缩得棕色油状物8 g,直接用于投下一步反应,收率85.1%。ESI-MS m/z: 366.1 [M+H]+。1H NMR (400 MHz, DMSO-d6):δ8.94 (t,J=6.2 Hz, 1H), 7.69~7.62 (m, 2H), 7.60~7.48 (m, 3H), 7.35 (ddd,J=9.0, 7.9, 3.3 Hz, 1H), 7.19 (dd,J=9.1, 4.3 Hz, 1H), 4.61 (d,J=6.1 Hz, 2H), 3.90 (d,J=2.2 Hz, 6H).
2.2.4 N-((4-(5-氨基-4-氰基-1-((1S)-2,2,2-三氟-1-甲基乙基)-1H-吡唑-3-基)苯基)甲基)-5-氟-2-甲氧基苯甲酰胺(X7)的制备
冰浴条件下将(1,1,1-三氟丙烷-2-基)肼盐酸盐(77 mg,0.59 mmol)溶于EtOH(2 mL)溶液中,滴加TEA(55 mg,0.54 mmol),搅拌30 min,然后再将上述溶液滴加入X6(200 mg,0.54 mmol)的EtOH溶液中,将混合物移至室温下搅拌反应12 h。TLC监测原料反应完毕。溶液冷却到0 ℃,抽滤,固体用EtOH洗3次,置于真空干燥箱干燥过夜,得白色固体200 mg,收率80.3%。ESI-MS m/z: 462.1 [M+H]+。1H NMR (400 MHz, DMSO-d6):δ8.84 (t,J=6.1 Hz, 1H), 7.75 (d,J=8.0 Hz, 2H), 7.51 (dd,J=9.2, 3.3 Hz, 1H), 7.43 (d,J=8.0 Hz, 2H), 7.33 (td,J=8.5, 3.3 Hz, 1H), 7.18 (dd,J=9.1, 4.3 Hz, 1H), 7.09 (s, 2H), 5.30 (p,J=6.7 Hz, 1H), 4.53 (d,J=6.1 Hz, 2H), 3.89 (s, 3H), 1.65 (d,J=6.8 Hz, 3H).
2.2.5 LOXO-305(1)的制备
将X7(200 mg,0.43 mmol)加入甲基磺酸(MsOH)(2 mL)的溶液中,于85 ℃搅拌反应3 h。TLC监测原料反应完毕。用NaOH溶液调pH至11左右,有固体析出,抽滤,固体用水洗3次,置于真空干燥箱干燥过夜,得黄色固体120 mg,收率(58.2%),纯度为99.5%[HPLC面积归一化法,色谱柱为Horizon C18柱(4.6 mm×250 mm,5 μm);流动相A为体积分数0.1%的三氟乙酸水溶液,流动相B为乙腈(梯度洗脱:0~1 min B 5%~20%, 1~11 min B 20%~40%, 11~11.2 min B 40%~95%, 11.2~13.2 min B 95%, 13.2~13.5 min B 95%~5%, 13.5~15 min B 5%);检测波长:254 nm;流速:1.0 mL·min-1;柱温:40 ℃]。ESI-MS m/z: 480.1[M+H]+。1H NMR (400 MHz, DMSO-d6):δ8.84 (t,J=6.1 Hz, 1H), 7.51 (dd,J=9.2, 3.3 Hz, 1H), 7.44 (q,J=8.1 Hz, 4H), 7.37~7.30 (m, 1H), 7.18 (dd,J=9.1, 4.3 Hz, 1H), 6.67 (s, 2H), 5.29 (p,J=7.2 Hz, 1H), 4.55 (d,J=6.1 Hz, 2H), 3.89 (s, 3H), 1.61 (d,J=6.8 Hz, 3H).
3 结果与讨论
3.1 中间体X3的方法优化
在合成化合物X3时,原研路线分为两步,先使用二氯亚砜作酰化试剂,将中间体制成酰氯,随后再与4-氨甲基苯甲酸偶联得到中间体X3,反应过程中既要控水,还要氮气保护,操作烦琐,而且受4-氨甲基苯甲酸溶解度的影响,总反应时长达20 h,且存在原料剩余问题。本工艺路线改用低成本的CDI缩合剂,两原料物质的量比为1∶1时一步反应即可得到中间体X3,室温下仅3 h即可反应完全,且不存在原料剩余问题,大大缩短了反应时间,收率提高至98.2%,后处理只要调节pH至3~4,减压浓缩一定量的溶剂,过滤干燥即得化合物X3,此步反应后处理避免了打浆操作,在保证纯度和收率的同时,简化了操作,更符合工业生产的要求。
3.2 中间体X5的方法优化
在合成化合物X5时,原研路线分为两步,先使用二氯亚砜作酰化试剂,将中间体制成酰氯,随后再与丙二腈偶联得到中间体X5,反应过程中既要控水,还要氮气保护,操作烦琐,总反应时长达20 h;本工艺路线改用缩合剂EDCI,一步反应即可得到中间体X5,室温下仅6 h即可反应完全,大大缩短了反应时间,收率提高至92.1%,后处理只要用碱水和酸水洗去反应体系中的HOBT,即得化合物X5,在保证纯度和收率的同时,简化了操作,更符合工业生产的要求。
3.3 中间体X6的方法优化
在合成化合物X6时,原研路线需要在92 ℃反应18 h;本工艺路线将温度提高至100 ℃,反应时长降低至10 h,很大程度上减少了反应时间,后处理只要旋蒸去除TMOF,直接投下一步反应即可,在保证后一步收率的同时,简化了操作。
4 结论
本文改进了LOXO-305的合成路线,以5-氟-2-甲氧基苯甲酸为起始原料,经两步简单的缩合反应得到关键中间体X5,很大程度地减少了反应时长,降低了合成成本。中间体X5经甲基化、关环反应、水解反应得到目标化合物LOXO-305,纯度为99.5%,改进后的工艺路线总收率为35.9%。该方法使用的试剂简单易得,后处理操作简便,产物纯化多采用水洗,避免柱层析分离,便于放大生产,可为LOXO-305及其衍生物的合成提供参考。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
云南化工(2020年11期)2021-01-14 00:50:52
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
铜仁学院学报(2018年6期)2018-07-05 09:47:36
中成药(2017年4期)2017-05-17 06:09:50
百科知识(2016年22期)2016-12-24 21:07:25
当代化工研究(2016年5期)2016-03-20 16:21:34
无机化学学报(2014年12期)2014-02-28 17:34:01
无机化学学报(2014年8期)2014-02-28 17:32:46
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:32
