
三维乒乓菊状CdS/BiOBr催化剂的制备及其光催化降解罗丹明B
2023-10-31 08:11李红颖任瑞鹏吴丽杰吕永康
纺织学报 2023年9期
李红颖, 徐 毅, 杨 帆, 任瑞鹏, 周 全, 吴丽杰, 吕永康
(1. 太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室, 山西 太原 030024;2. 太原理工大学 化学工程与技术学院, 山西 太原 030024)
随着人口的日益增加,能源供应和环境污染成为不可回避的严峻考验[1]。太阳能的高效利用对人类社会具有深远影响。在太阳光中,可见光所占比例为45%左右,可见光的有效利用是太阳能源利用的重要组成部分。随着我国纺织工业的持续快速发展,由印染废水所引起的环境问题受到了越来越多的关注[2]。罗丹明B(RhB)、甲基橙(MO)等具有发色团的含氮染料具有稳定的化学性质,会使水体颜色加深影响透光率,且具有致癌和导致基因突变等危害[3-4]。针对废水中的染料,常用生物降解法、电化学法、化学氧化法、混凝法、吸附法等进行处理[5],但这些方法往往存在局限性。作为一种成本低且绿色环保的高级氧化技术,光催化技术利用太阳能产生强氧化性的活性物质,将有机染料氧化降解为无毒小分子物质,在染料废水处理技术中显现出独特的优势[6]。
卤氧化铋(BiOX,X可为F、Cl、Br和I)具有稳定的化学性质,无毒且抗腐蚀,作为一种新型的层状半导体材料[7-8],受到了越来越多学者的关注。其中BiOBr具有良好的可见光响应,越来越多的研究报道了BiOBr在光催化方面的应用[9-11]。然而,纯组分BiOBr虽然具备一定的光催化性能,但其禁带宽度稍大,可见光利用率偏低,光生载流子易发生复合。在以往的研究中,研究者通过构建异质结,调整微观结构,元素掺杂等方法对单组分BiOBr进行改进[12]。其中,通过异质结的构建可使光生电子-空穴有效分离,进而达到提高光催化活性的目的。已有许多研究合成出了具有良好可见光响应的BiOBr基半导体异质结[13-15]。CdS是一种重要的II-IV族半导体,在光催化降解领域有着广泛的应用[16-18]。CdS由于较窄的禁带宽度,已被广泛用作敏化剂,以改善宽带隙半导体的可见光响应[19]。光生空穴自氧化引起的光腐蚀对其稳定性不利[20],通过与其它半导体复合可以促进光生电子-空穴分离,从而提高光催化性能,同时CdS价带中光生空穴发生转移能够有效抑制CdS的光腐蚀[21]。BiOBr和CdS具有匹配良好的能带位置,二者可复合形成异质结构。其中,CdS可作为可见光敏化剂,增强催化剂对可见光的吸收。BiOBr和CdS形成异质结后,能够减少光生电子-空穴的复合,提高光催化效率,同时可以抑制CdS的光腐蚀。目前已有部分文献报道过CdS/BiOBr异质结在可见光下降解染料的研究。
Liu等[22]将制备好的CdS和BiOBr微球分散在溶液中,通过溶剂热法得到CdS量子点敏化的BiOBr微球,该微球在降解甲基橙(MO)实验中表现出较好的降解活性,但循环4次后降解率下降至初始值的85%左右。牟小冬等[23]通过化学沉积法制备了CdS/BiOBr复合微球,光照120 min后,RhB的光降解率为85%。Cui等[24]利用沉淀法合成了CdS/BiOBr催化剂,该催化剂表现出优异的RhB光降解活性及循环稳定性,但CdS颗粒在BiOBr纳米球表面分布无规则。You等[25]通过两步超声波-沉淀法合成的CdS/BiOBr纳米催化剂在降解RhB过程中表现出较强的降解活性,但催化剂的循环稳定性不好、重复利用率欠佳,在光催化降解5个循环后降解率仅为50%左右。以上研究均存在同一个问题,CdS/BiOBr催化剂中CdS和BiOBr进行无规则组合,复合催化剂的活性或稳定性不好。
本文采用两步法合成了一种三维乒乓菊状CdS/BiOBr催化剂,首先利用溶剂热法制备了BiOBr纳米片,然后采用水热法直接在层状BiOBr表面生长CdS粒子制备了CdS/BiOBr催化剂,通过调整CdS和BiOBr的量比来调控催化剂的形貌;并通过一系列表征手段分析了催化剂的组成、形貌以及光学性能等特征,在可见光照射下考察了材料的光催化活性,通过循环利用实验、活性组分检测探讨了催化剂的稳定性和光催化机制。
1 材料和方法
1.1 实验药品
五水硝酸铋(Bi(NO3)3·5H2O)、九水硫化钠(Na2S·9H2O),购自麦克林生化科技有限公司;乙二醇(EG)、无水乙醇(EA),购自大茂化学试剂厂;溴化钾(KBr),购自致远化学试剂有限公司;二水醋酸镉(Cd(Ac)2·2H2O)、罗丹明B(RhB)、对苯醌(BQ)、叔丁醇(TBA)、乙二胺四乙酸(EDTA),均购自阿拉丁生化科技股份有限公司;实验用水为超纯水。所有实验药品均为分析级,且无需进一步纯化。
1.2 催化剂的制备
1.2.1 BiOBr的制备
纯组分BiOBr按照文献[1]报道的溶剂热法进行制备。
1.2.2 CdS/BiOBr催化剂制备
采用水热法制备了不同量比的CdS/BiOBr催化剂。本文以制备CdS与 BiOBr的量比为1∶1(nC∶nB=1∶1)的催化剂为例进行介绍,称量0.003 mol Cd(Ac)2·2H2O溶解在70 mL超纯水中,搅拌20 min,再超声波溶解10 min。之后将0.003 mol BiOBr加入到上述溶液中,搅拌10 min。随后称量0.003 mol Na2S·9H2O加入上述溶液,搅拌20 min,超声波处理10 min。最后将溶液转移到100 mL水热反应釜中,在140 ℃反应8 h。反应结束后自然冷却至室温,用超纯水和无水乙醇清洗3次,在60 ℃烘箱中干燥12 h。其它量比的催化剂也按照上述方法制备,相同条件下不添加BiOBr制备纯组分CdS。选择CdS与BiOBr的量比分别为5∶1、3∶1、1∶1、1∶3、1∶5制备5种量比的催化剂CdS/BiOBr(5∶1)、CdS/BiOBr(3∶1)、CdS/BiOBr(1∶1)、CdS/BiOBr(1∶3)、CdS/BiOBr(1∶5)。图1为CdS/BiOBr催化剂的制备示意图。

图1 CdS/BiOBr催化剂的制备示意图Fig. 1 Schematic diagram of preparation of CdS/BiOBr catalyst
1.3 催化剂的表征
1.3.1 样品形貌测试
采用德国蔡司公司ZEISS Sigma 300扫描电子显微镜和JEM-2100F透射电子显微镜表征样品的形貌和结构。透射电子显微镜测试前,在乙醇中溶解少量粉末样品,进行超声波分散,利用毛细滴将样品的分散液管滴加到超薄碳膜上,最后真空干燥后进行测试。
1.3.2 晶体结构测试
采用日本理学公司Rigaku MiniFlex 600 X射线衍射(XRD)仪表征样品的晶体结构。采用CuKα射线(λ=0.154 18 nm)进行样品辐射,2θ范围为5°~80°,扫描速度为10(°)/min。
1.3.3 紫外-可见漫反射光谱测试
采用日本岛津公司UV-3600i Plus紫外-可见漫反射光谱(UV-Vis DRS)分析仪表征样品的光学性能,样品测试范围为200~800 nm。样品的带隙能Eg由下式[26]计算:
αhν=A(hν-Eg)n/2
(1)
式中:α为光学吸收系数;h为普朗克常数;ν为光频率,Hz;Eg为带隙能,eV;A为比例常数。BiOBr和CdS的n值分别选择4和1。利用以下经验公式[27]可计算催化剂的价带(VB)和导带(CB)的位置:
EVB=X-Ee+0.5Eg
(2)
ECB=EVB-Eg
(3)
式中:EVB为价带电位;ECB为导带电位;X为电负性,BiOBr和CdS的X值分别为6.18和5.05 eV;Ee为在氢尺度上的自由电子能量,约为4.5 eV。
1.3.4 电子-空穴复合率测试
采用英国爱丁堡公司Edinburgh FLS1000稳态/瞬态荧光光谱仪测试光致发光光谱(PL),用于表征样品电子-空穴复合率,激发波长为325 nm。
1.3.5 表面性能分析
采用美国安捷伦公司ASIQ 050200-6 BET表面分析仪测试样品的比表面积。预处理条件为真空条件下150 ℃处理8 h。将预处理后的样品在-196 ℃的液氮中冷却,测得样品的吸脱附曲线。利用Brunauer-Emmet-Teller (BET)计算催化剂的比表面积。
1.3.6 电子自旋共振测试
采用德国Bruker公司 EMXplus-6/1电子自旋共振谱仪测试光催化降解过程中是否产生·O2-、h+。·O2-的捕获剂为5,5-二甲基-1-吡咯啉-N-氧化物(DMPO),以2,2,6,6-四甲基哌啶-1-氧自由基(TEMPO)作为h+的捕获剂,DMPO浓度为100 mmol/L,TEMPO浓度为100 mmol/L。测试·O2-时以甲醇为溶剂,测试h+时以去离子水作溶剂。
1.4 光催化剂的吸附能力
在黑暗中建立光催化剂与RhB分子之间的吸附-解吸平衡。催化剂对RhB的吸附量qe(mg/g)为
qe=(C1-C2)V/m
(4)
式中:C1为溶液中RhB的初始浓度,mg/L;C2为平衡浓度,mg/L;V为溶液体积,L;m为干催化剂的质量,g。
1.5 光催化性能实验
通过RhB在可见光照射下的降解来评价样品的光催化性能。实验中可见光源为500 W氙灯。将0.03 g光催化剂分散于100 mL RhB(20 mg/L)溶液中。在光照前,将悬浮液在黑暗中机械搅拌1 h,建立光催化剂和RhB分子之间的吸附-解吸平衡。在光催化活性实验过程中,经过一定时间取3.0 mL悬浮液,然后将粉末状样品离心分离,采用SP-752紫外可见分光光度计在554 nm下测定RhB水溶液的吸光度以计算其浓度。RhB的降解率y为
(5)
式中:C0为RhB溶液的初始质量浓度,mg/L;Ct为时间t时B溶液的质量浓度,mg/L。
1.6 动力学常数计算
采用Langmuir-Hinshelwood(L-H)方程来描述光催化降解RhB的动力学,揭示了初始浓度与降解速率常数之间的关系。
(6)
式中:k0为拟一阶速率常数,min-1;kini表示固有速率常数,mmol/(L·min);KRhB为RhB在样品表面的L-H吸附常数,mmol/(L·min)。
式(6)可简化为一级反应动力学:
-ln(Ct/C0) =kiniKRhBt=Kt
(7)
式中,K为反应速率常数。
1.7 稳定性测试
通过光催化循环实验评估催化剂的稳定性。催化剂的循环利用实验与光催化活性评价实验过程相似,每次光催化活性测试后,对催化剂离心分离,洗涤后在60 ℃下干燥24 h,再用于下一次循环实验。
1.8 活性物种捕获实验
有机污染物的降解取决于光催化剂表面产生的活性物质。在光催化反应过程中产生的e-、h+、·O2-或·OH能与吸附在光催化剂表面的RhB分子快速反应。实验通过BQ捕获·O2-、EDTA捕获h+、TBA捕获·OH。活性物种捕获实验与光催化活性评价实验过程相同,通过添加3种捕获剂以对比不添加捕获剂情况下的动力学常数变化。
2 结果与讨论
2.1 形貌分析
图2为不同样品的扫描电镜照片。从图2(a)、(h)可看出,纯组分CdS为均匀的纳米球,BiOBr为规则的纳米片。从图2(b)~(g)可看出,CdS/BiOBr催化剂包括CdS和BiOBr两相,通过调节二者的含量其微观形貌发生了改变。当CdS和BiOBr 的量比为5∶1时,CdS含量较多,BiOBr和CdS类似机械混合,BiOBr片状结构被CdS覆盖。当CdS和BiOBr的量比为3∶1时,其形貌发生变化,片状结构开始出现团聚,CdS颗粒覆盖在BiOBr片状结构表面且呈现棉絮状。进一步降低CdS的含量至二者量比为1∶1时,呈现规律的自组装层级结构,且CdS粒子紧密负载在层状BiOBr表面。如图2(e)、(f)所示,当CdS与BiOBr的量比为1∶3时,催化剂发生明显的形貌变化,呈现三维乒乓菊状层级结构,此时CdS纳米颗粒均匀地分布在BiOBr花瓣状薄片的表面。该结构具有大量的间隙,使光更容易照射到催化剂内部,为光反应提供了足够的传输路径,使界面光生载流子有效转移,从而提高光催化剂的催化性能。但随着CdS含量继续降低,三维乒乓菊状结构消失,二者又呈现出机械混合,在BiOBr纳米片附近观察到团聚的CdS纳米颗粒。

图2 CdS及不同量比CdS/BiOBr催化剂和BiOBr的SEM照片Fig. 2 SEM images of CdS, different ratios of CdS/BiOBr catalysts and BiOBr
图3(a)示出CdS/BiOBr(1∶3)催化剂的EDS光谱,可观察到与Bi、Br、O、Cd和S相关的峰,证实CdS/BiOBr催化剂中这6种元素的存在。图3(b)、(c)示出CdS/BiOBr(1∶3)催化剂的高分辨率透射电镜照片。计算得到CdS晶格条纹的平面晶格间距为0.205 nm,与CdS(220)晶格面相吻合;BiOBr晶格条纹的平面晶格间距为0.275 nm,与BiOBr(110)晶格面相吻合。进一步证明CdS和BiOBr在CdS/BiOBr催化剂中形成异质结。

图3 CdS/BiOBr(1∶3)催化剂的EDS图像和TEM照片Fig. 3 EDS image(a)and TEM image (b)of CdS/BiOBr (1∶3) catalyst
2.2 晶体结构分析
图4示出CdS,BiOBr和CdS/BiOBr(1∶3)催化剂的XRD图谱。在纯组分BiOBr的衍射图谱中,31.7°、32.2°、46.2°、57.2°等处出现分别对应于BiOBr(102)、(110)、(020)、(212)晶面的衍射峰,与四方相结构BiOBr(JCPDS No.73-2061)一致[28],并且其衍射峰强烈而清晰,说明制备的BiOBr结晶度良好。制备的纯组分CdS在26.5°、43.97°、52.08°处出现明显的CdS(111)、(220)、(311)晶面衍射峰,与CdS(JCPDS No.75-1546)[29]一致,样品显示出宽而弱的峰,表明化合物的非晶态性质或样品的纳米晶行为。在上述单组分衍射峰位置,CdS/BiOBr催化剂同样出现了衍射峰,且无其它杂质衍射峰,表明CdS/BiOBr异质结成功制备且纯度较高。与纯组分BiOBr相比,CdS/BiOBr催化剂中BiOBr的衍射峰明显减弱,这表明CdS成功沉积到BiOBr表面形成CdS/BiOBr异质结。CdS/BiOBr催化剂中CdS的衍射峰相对较弱,出现这种结果的原因一方面是CdS在CdS/BiOBr催化剂中含量较少,另一方面是CdS单体本身的衍射峰相对较弱。

图4 CdS,CdS/BiOBr(1∶3)催化剂和BiOBr的XRD图谱Fig. 4 XRD patterns of CdS, CdS/BiOBr (1∶3) catalyst and BiOBr
2.3 紫外-可见漫反射分析
图5示出样品的紫外-可见漫反射光谱图及禁带宽度图。如图5(a)所示,纯BiOBr和纯CdS的吸收带边缘约为440和560 nm。CdS/BiOBr(1∶3)催化剂观察到2个吸收带边缘,分别归因于纯CdS和CdS/BiOBr催化剂[30]。与BiOBr和CdS相比均发生了明显的红移,此外,CdS/BiOBr(1∶3)催化剂在550~800 nm区域的吸收强度显著增强,因此,CdS/BiOBr催化剂对可见光的吸收增强,能产生更多的光生载流子,有利于光催化性能的提高。如图5(b)、(c)所示,BiOBr和CdS的禁带宽度分别为2.70和 2.28 eV。根据式(2)、(3),计算得到BiOBr的CB和VB位置分别为0.33和3.03 eV,CdS的CB和VB位置分别为-0.59和1.69 eV。说明BiOBr和CdS能够形成良好的复合结构,有利于光生载流子的分离。

图5 紫外-可见漫反射光谱及禁带宽度图Fig. 5 UV-Vis diffuse reflectance spectrum and forbidden band width. (a) UV-Vis diffuse reflectance spectroscopy; (b) Forbidden band width of BiOBr; (c) Forbidden band width of CdS
2.4 电子-空穴复合率分析
图6为样品的PL图谱。可看出,BiOBr的荧光强度较强,其电子-空穴的复合率较高。与BiOBr相比,三维乒乓菊状CdS/BiOBr(1∶3)催化剂的荧光光谱强度明显减弱,原因是CdS/BiOBr异质结的形成可使光生电子-空穴有效分离。

图6 BiOBr和CdS/BiOBr(1∶3)催化剂的PL图谱Fig. 6 PL profiles of BiOBr and CdS/BiOBr (1∶3)catalyst
2.5 表面性能分析
图7示出样品的等温线。可看出样品的等温线均表现为典型的Ⅳ类型曲线,说明制备的催化剂为介孔材料。测试结果显示纯组分 BiOBr的比表面积为15.52 m2/g,CdS的比表面积为130.66 m2/g,三维乒乓菊状CdS/BiOBr(1∶3)催化剂的比表面积较纯组分BiOBr增大,为33.85 m2/g。比表面积的增大增强了复合催化剂的吸附能力,且较大的比表面积有利于催化剂对光的吸收,并为催化剂降解污染物提供足够的反应场所。

图7 CdS,CdS/BiOBr(1∶3)催化剂和BiOBr的氮气吸-脱附曲线Fig. 7 Nitrogen adsorption-desorption curves for CdS, CdS/BiOBr (1∶3) catalyst and BiOBr
2.6 光催化降解RhB性能
图8为不同样品的RhB降解图、动力学曲线图及动力学常数图。图8(a)为BiOBr、CdS和不同量比CdS/BiOBr催化剂的光催化活性图。样品在可见光照射前,于黑暗中搅拌60 min,建立RhB与光催化剂之间的吸附-解附平衡。吸附平衡后,BiOBr,CdS以及不同量比的CdS/BiOBr催化剂对RhB的吸附率分别为12.3%、20.7%、17.1%、19.2%、22.2%、29.3%和23.5%。CdS/BiOBr(1∶3)催化剂的吸附率最大,归因于其较大的比表面积和独特的形貌结构。光照120 min后,BiOBr对RhB的降解率为75.3%,CdS对RhB的降解率为30.4%,不同量比的CdS/BiOBr催化剂的降解率分别为40.69%、60.28%、79.03%、99.5%、90.72%,其中CdS/BiOBr(1∶3)催化剂对RhB的降解率最大。光催化降解实验结果表明,CdS/BiOBr催化剂中CdS含量对其光催化性能有影响,当CdS和BiOBr的量比为1∶3时,CdS/BiOBr催化剂的光催化降解RhB的性能最好。光催化降解过程符合一级动力学模型。如图8(b)、(c)所示,CdS/BiOBr(1∶3)催化剂具有最大的降解速率常数K(0.044 min-1),分别是BiOBr和CdS速率常数的3.67倍和40倍。催化剂K值的顺序从大到小为CdS/BiOBr(1∶3)、CdS/BiOBr(1∶5)、CdS/BiOBr(1∶1)、BiOBr、CdS/BiOBr(3∶1),CdS/BiOBr(5∶1)、CdS。CdS/BiOBr(1∶3)催化剂优异的降解率与降解速率常数可归因于其较强的可见光吸收效率,较低的电子-空穴对复合率,而且独特的三维乒乓菊状层级结构间隙丰富,使光线更容易射入催化剂内部,该结构使反应分子具有更充足的传输路径,使界面光生载流子快速转移,进而增强光催化性能。

图8 不同样品的RhB降解图、动力学曲线及动力学常数图Fig. 8 Photocatalytic degradation of RhB (a), kinetic curves of RhB degradation (b) and primary kinetic constants of RhB degradation (c) of different samples
2.7 稳定性分析
通过7次光催化循环实验评估了光催化剂的稳定性。图9示出CdS/BiOBr(1∶3)催化剂的重复利用性能。可看出,三维乒乓菊状光催化剂在降解RhB循环7次后,其光催化活性仍保持良好,降解率由99.5%降至90.1%。

图9 CdS/BiOBr(1∶3)催化剂的重复利用性能Fig. 9 Reuse performance of CdS/BiOBr (1∶3)catalyst
7次光催化循环实验后,三维乒乓菊状催化剂的晶相结构和微观形貌通过XRD和SEM进行分析,结果如图10、11所示。如图10所示,催化剂的XRD衍射峰在循环使用前后没有发生明显的变化,说明其晶体结构基本没有被破坏。如图11所示,7次光催化循环实验后,催化剂的三维乒乓菊状形貌保持良好。这表明三维乒乓菊状催化剂具有良好的稳定性。

图10 CdS/BiOBr(1∶3)催化剂循环使用7次前后的XRD图谱Fig. 10 XRD patterns of CdS/BiOBr (1∶3)catalyst before and after 7 cycles

图11 CdS/BiOBr(1∶3)催化剂循环使用7次前后的SEM照片Fig. 11 SEM images of CdS/BiOBr (1∶3)catalyst before(a)and after(b) 7 cycles
考虑到Cd2+的毒性,使用原子吸收光谱仪测定了RhB光降解后溶液中Cd2+的含量。三维乒乓菊状CdS/BiOBr催化剂降解后,RhB溶液中Cd2+的质量浓度为5.02 mg/L。然而,在CdS光催化剂的情况下,相应的Cd2+质量浓度为43 mg/L。结果证实,CdS/BiOBr异质结的构建使CdS价带中的光生空穴转移到BiOBr,在一定程度上减少了CdS的光腐蚀[31],使催化剂在RhB的循环降解中表现出良好的稳定性。
2.8 活性物种捕获实验结果分析
通过对活性物种的捕获实验,研究了光催化降解RhB过程中的主要活性物质。图12示出捕获剂对CdS/BiOBr(1∶3)催化剂降解RhB的影响。可看出,向光催化降解系统中添加TBA后,RhB的降解速率常数略有降低,说明·OH对RhB光催化降解的影响相对较弱。然而在光催化体系中加入EDTA后,RhB的降解活性明显受到抑制,降解速率常数明显降低,说明h+是RhB降解过程中的重要活性物质。添加BQ对光催化剂活性的抑制作用最为明显,说明·O2-在光催化降解过程中起着关键作用。从以上分析可以推断,·O2-和h+在RhB降解过程中发挥了重要作用,这与文献[25]报道一致。

图12 捕获剂对CdS/BiOBr(1∶3)催化剂降解RhB的影响Fig. 12 Influences of trapping agents on degradation of RhB by CdS/BiOBr (1∶3)catalyst
2.9 电子自旋共振测试结果分析


图13 可见光下CdS/BiOBr(1∶3)催化剂EPR谱图Fig. 13 EPR spectra of CdS/BiOBr (1∶3) catalyst under visible light
2.10 光催化降解机制分析
图14示出可见光下CdS/BiOBr(1∶3)催化剂对RhB降解机制示意图。可看出,根据制备的CdS/BiOBr催化剂的带隙结构以及捕获剂的作用、EPR测试结果,提出了一种可能的光催化降解途径。当CdS/BiOBr催化剂受到可见光照射时,CdS和BiOBr的CB以及VB中分别产生光生电子和空穴。由于二者界面的紧密接触以及能带结构的良好匹配,CdS产生的光生电子转移到BiOBr的CB中,与此同时CdS的VB中富集了空穴,这个过程有效抑制了光生载流子的复合。由于CdS的CB(-0.59 eV)比·O2-的形成电位(-0.046 eV)更负[32],因此CdS的CB中产生的光生电子可以与O2分子结合生成·O2-,这个过程也可发生在CdS/BiOBr催化剂的接合界面处。而CdS的VB(1.69 eV)低于OH-/·OH(2.38 eV)的氧化还原电位[33],因此光生孔不会氧化OH-成为·OH,但CdS的VB中富集的光生空穴非常活跃,可直接将RhB分解[34],因此,CdS/BiOBr催化剂光降解RhB过程中的主要活性自由基是·O2-以及CdS价带中富集的光生空穴。这与活性物质捕获结果及EPR测试结果一致。
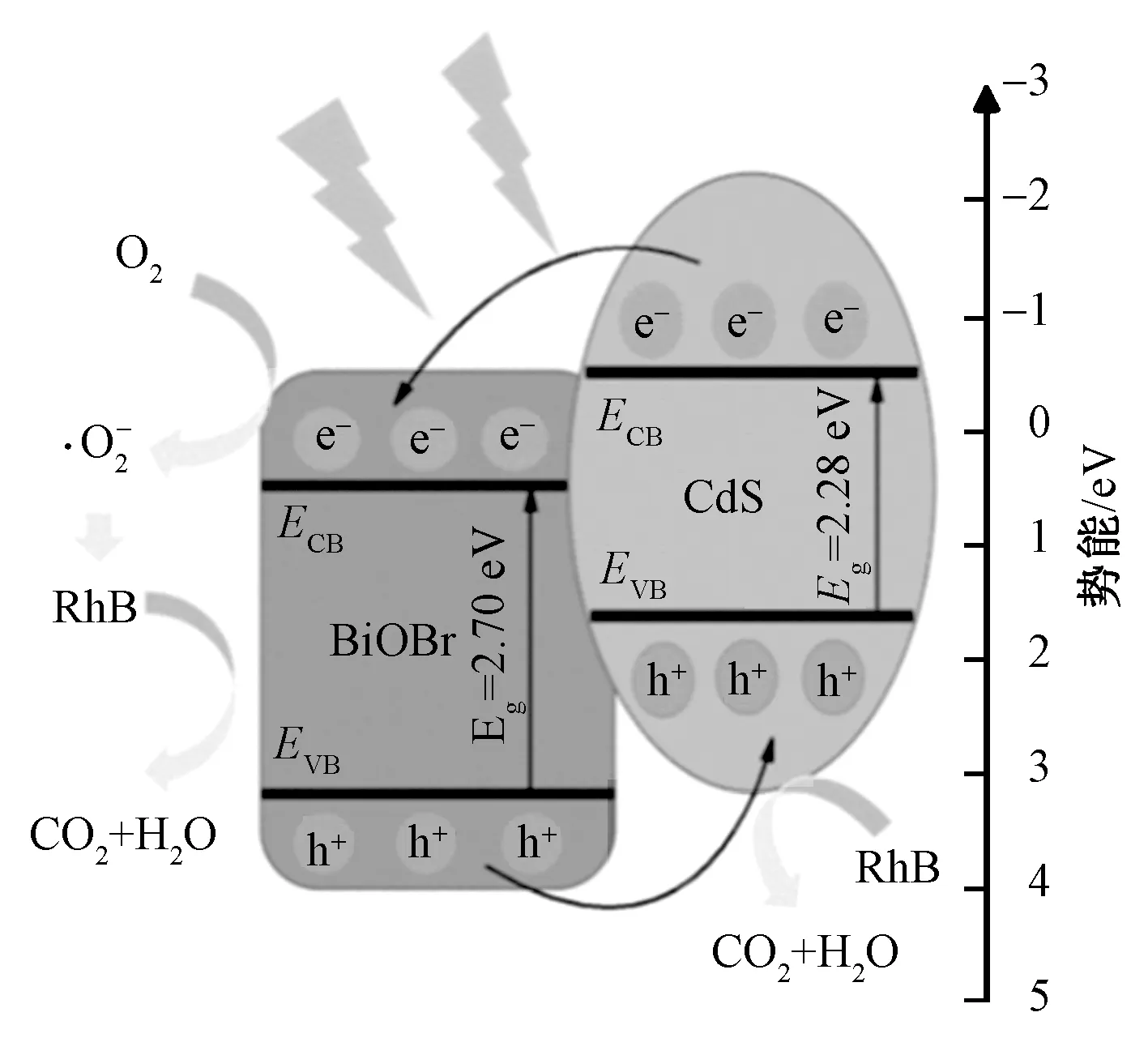
图14 可见光下CdS/BiOBr(1∶3)催化剂对RhB降解机制示意图Fig. 14 Schematic diagram of RhB degradation mechanism on CdS/BiOBr (1∶3) catalyst under visible light
3 结 论
本文合成了一种规则组合的三维乒乓菊状CdS/BiOBr催化剂,通过各种表征测试证实该催化剂具备良好的晶体结构、光学性能等。在降解罗丹明B的过程中,表现出优异的光催化活性和循环稳定性。这归因于,制备的CdS/BiOBr催化剂显示出比CdS和BiOBr更好的可见光吸收效率,更低的电子-空穴对复合概率,而且三维乒乓菊状层级结构比表面积更大,提供了充足的反应传输场所,使界面光生载流子快速转移,进而提高催化效率。活性物种捕获实验和电子自旋共振测试确定了催化过程中·O2-和h+是主要活性物种。
猜你喜欢
车用发动机(2021年5期)2021-10-31
电源技术(2021年7期)2021-07-29
——潘桂棠光生的地质情怀
沉积与特提斯地质(2021年2期)2021-07-20
陶瓷学报(2019年6期)2019-10-27
赤峰学院学报·自然科学版(2018年7期)2018-08-11
物理化学学报(2017年3期)2017-03-11
浙江农业科学(2016年11期)2016-05-04
昭通学院学报(2016年5期)2016-02-24
石家庄铁道大学学报(自然科学版)(2015年3期)2015-02-28
应用化工(2014年11期)2014-08-16
