
藏药六味石榴丸的质量标准研究
2023-10-25 12:13周德来龙布才让张明童
中国民族民间医药 2023年19期
李 运 周德来 龙布才让 张明童
1.兰州市食品药品检验检测研究院,甘肃 兰州 730050;2.甘肃中医药大学药学院,甘肃 兰州 730000;3.甘南州合作市卡加曼藏药开发有限公司,甘肃 合作 747000;4.甘肃省药品检验研究院,甘肃 兰州 730070
六味石榴丸源于《四部医典》,藏文名“赛知周巴”,由石榴子100 g、肉桂50 g、肉豆蔻50 g、胡椒50 g、红花50 g、大托叶云实50 g等六味药材组成[1]。方中石榴子治一切胃病,能提升胃阳,并且可治疗培根寒证;肉桂散热止痛、温通筋脉;肉豆蔻温中行气;胡椒温中散寒;红花活血通经、散瘀止痛;大托叶云实温肾,逐寒等,诸药共奏调节三因紊乱,温肾、暖腰膝之功效。临床常用于妇女白带病。该方始源于人们实践的直接经验,有较牢靠的经验基础,在指导数千年医疗实践活动中,得到不断创新完善。为了提高药效的持久性,减少吸潮,便于贮存,并增加服用及携带的便利性,以六味石榴散为基础,将其剂型改变提升为丸剂(水丸),并建立较完善的质量标准,以保障药品质量。课题组在前期工作的基础上,结合相关文献[2-9]研究,建立了处方中主要原料的显微鉴别、薄层鉴别方法,并采用 HPLC 法分别对制剂中石榴子所含鞣花酸、红花中羟基红花黄色素A的含量进行测定,从而更加有效、系统地控制藏药六味石榴丸的质量,确保临床用药安全、有效。
1 仪器与材料
1.1 仪器 BX53+DP73正置荧光生物显微镜(日本奥林巴斯株式会社);薄层色谱成像系统(瑞士CAMAG公司);Waters e2695高效液相色谱仪(美国Waters公司);XSE205DU型分析天平(瑞士Mettle Toledo公司);SB-800DT型超声波清洗机(宁波新芝产品)。
1.2 材料 石榴子对照药材(批号:121431-201002)、肉豆蔻对照药材(批号:120926-201608)、红花对照药材(批号:120907-201713)、胡椒碱对照品(批号:110775-202107,纯度:98.2%)、鞣花酸对照品(批号:111959-201903,纯度:88.8%)、羟基红花黄色素A对照品(批号111637-202111,纯度:96.8%)均由中国食品药品检定研究院提供。六味石榴丸(批号:210801、210802、210803)由甘南州合作市卡加曼藏药开发有限公司生产,阴性样品由实验室模拟制得;硅胶G、硅胶GF254、硅胶H薄层板(规格:10 cm×10 cm,默克公司);甲醇、乙腈(色谱纯),水为Milli-Q超纯水,其他试剂为分析纯。
2 方法与结果
2.1 显微鉴别 取六味石榴丸粉末适量,进行显微制片(3~5张),置镜下观察可见:石细胞无色,椭圆形或类圆形,壁厚,孔沟细密(石榴子)。石细胞呈类圆形或类长方形,直径约32~80 μm,一面壁菲薄(肉桂)[2,10]。石细胞淡黄色,成群或散在,呈类圆形或多角形,直径20~35 μm,胞腔大,壁厚,木化,孔沟明显(胡椒)。花粉粒圆球形或椭圆形,直径约60 μm,外壁有刺,具三个萌发孔(红花)。结果如图1。

1.花粉粒(红花);2.分泌细胞(红花);3.石细胞(石榴子);4.石细胞(胡椒);5.石细胞(肉桂)
2.2 定性鉴别
2.2.1 石榴子的薄层色谱鉴别 取六味石榴丸粉末约1.0 g,置离心管中,加入乙醇溶液10 mL,超声提取30 min,取其上清液作为供试品溶液;另取石榴子对照药材1.0 g,按上述方法制备对照药材溶液。依据处方配比及制备方法,完成缺石榴子阴性对照样品的制备,照供试品溶液的制备方法,同法制成阴性对照溶液。吸取上述供试品及阴性对照溶液各5 μL,对照药材溶液4 μL,点于同一硅胶GF254薄层板上,以石油醚(60~90 ℃)-乙酸乙酯(9∶1)作为薄层展开系统,展开,取出,晾干,置紫外灯(254 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,应显相同颜色的荧光猝灭斑点,且与阴性对照样品中不存在干扰[11],如图2所示。
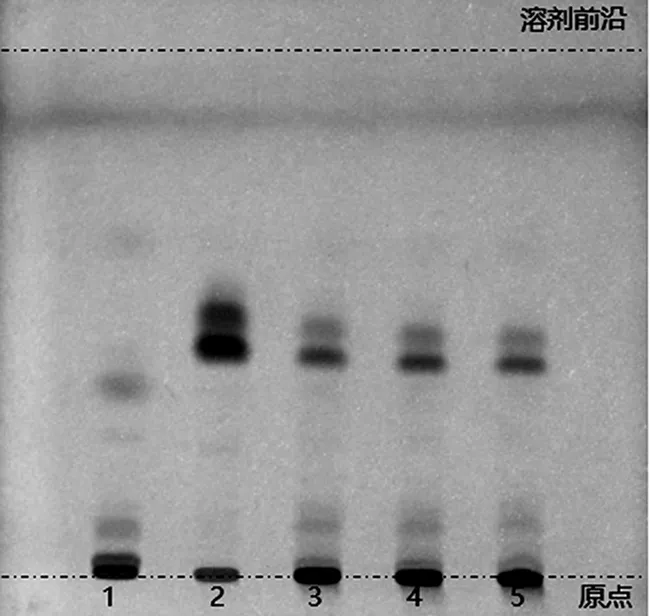
1.阴性对照样品溶液;2.对照药材溶液;3~5.供试品溶液
2.2.2 肉豆蔻的薄层色谱鉴别 取六味石榴丸粉末约1.0 g,置离心管中,加入石油醚(60~90 ℃)10 mL,超声提取30 min,滤过,取续滤液作为供试品溶液。另取肉豆蔻对照药材1.0 g,按上述方法制备对照药材溶液。依据处方配比及制备方法,完成缺肉豆蔻阴性对照样品的制备,照供试品溶液的制备方法,同法制成阴性样品溶液。吸取上述溶液各5 μL,分别点于同一高效硅胶G预制薄层板上,以石油醚(60~90 ℃)-乙酸乙酯(9∶1)作为薄层展开系统,经预饱和后,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105 ℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,应显相同颜色的斑点,且阴性对照样品中不存在干扰[11],如图3所示。
1.阴性对照样品溶液;2.对照药材溶液;3~5.供试品溶液
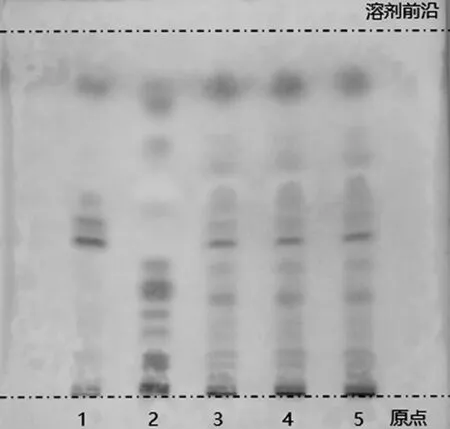
2.2.3 胡椒的薄层色谱鉴别 取六味石榴丸粉末约1.0 g,置离心管中,加入无水乙醇10 mL,超声提取30 min,滤过,将续滤液作为供试品溶液。另取胡椒碱对照品,置棕色容量瓶中,加无水乙醇制成每1 mL含2 mg的溶液,作为对照品溶液。按处方比例及制备工艺,制备缺胡椒的阴性对照样品,照供试品溶液的制备方法,同法制成阴性对照样品溶液。吸取供试品及阴性对照样品溶液各4 μL,对照品溶液2 μL,分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯-丙酮(7∶2∶1)作为薄层展开系统,展开,取出,晾干,喷以10%硫酸乙醇溶液,加热至斑点显色清晰,分别置日光和紫外光灯(365 nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,应显相同颜色的斑点或荧光斑点,且阴性对照样品中不存在干扰,如图4所示。
1.阴性对照样品溶液;2~4.供试品溶液;5.对照品溶液;A.日光下检视;B.紫外光灯(365 nm)下检视
2.2.4 红花的薄层色谱鉴别 取六味石榴丸粉末约1.0 g,置离心管中,加入80%丙酮溶液10 mL,密塞,振摇15 min,静置后取上清液,作为供试品溶液。另取红花对照药材0.5 g,加入80%丙酮溶液5 mL,同法制成对照药材溶液。依据处方配比及制备方法,完成缺红花阴性对照样品的制备,照上述供试品溶液的制备方法,同法制成阴性对照样品溶液。吸取上述溶液各5 μL,分别点于同一硅胶H薄层板上,以乙酸乙酯-甲醇-甲酸-水(7∶0.4∶2∶3)作为薄层展开系统,展开,取出,晾干。供试品色谱中,在与对照药材色谱相应的位置上,应显相同颜色的斑点,且阴性对照样品中不存在干扰,如图5所示。

1.阴性对照样品溶液;2.对照药材溶液;3~5.供试品溶液
2.3 六味石榴丸中鞣花酸的HPLC含量测定
2.3.1 对照品溶液的配置 精密称取鞣花酸对照品12.74 mg,置于25 mL容量瓶,用甲醇溶解、定容,摇匀。另取1 mL至10 mL容量瓶中,用甲醇定容至刻线即得对照品储备液,备用。
2.3.2 供试品溶液的制备 取六味石榴丸适量研细,另取约1.0 g,精密称定,置具塞锥形瓶中,精密加入甲醇50 mL,称定重量,超声提取(功率 300 W,频率40 kHz)50 min,放冷,再称定重量,用甲醇补足减失的重量,摇匀,静置后,取上清液经0.22 μm微孔滤膜滤过,取续滤液作为供试品溶液。
2.3.3 阴性对照样品溶液的制备 依据处方配比及制备方法完成不含石榴子药材的阴性对照样品的制备,按照“2.3.2”项下方法制备阴性对照样品溶液。
2.3.4 色谱条件 CAPCELL PAK-C18(250 mm×4.6 mm,5 μm);柱温:30 ℃;以乙腈-0.7%磷酸溶液(16∶84)为流动相;流速:1.0 mL/min;检测波长:252 nm。理论板数按鞣花酸峰计算应不低于4000。

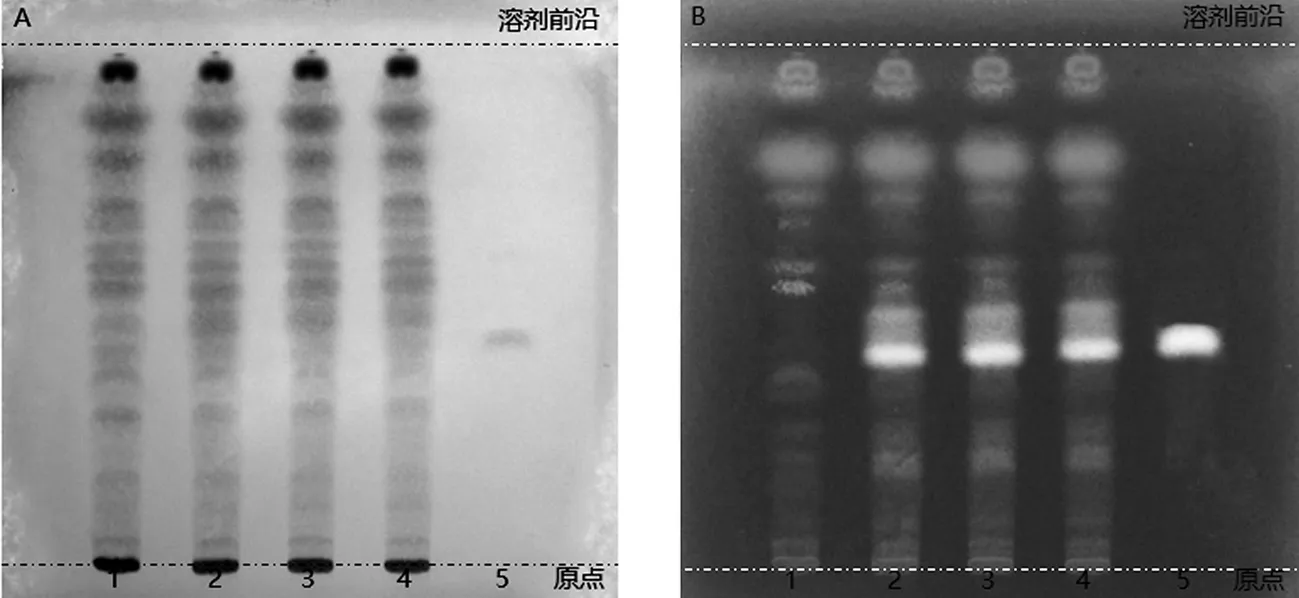
A.对照品溶液;B.供试品溶液;C.阴性对照样品溶液;1.鞣花酸

A.对照品溶液;B.供试品溶液;C.阴性样品溶液;1.羟基红花黄色素A
2.3.5 专属性试验 取上述对照品溶液10 μL、供试品溶液和阴性对照样品溶液,各20 μL,按照“2.3.4”项下条件检测。由图6可见,目标化合物分离度良好;阴性样品色谱图中,鞣花酸对应目标化合物在相应的保留时间无色谱峰吸收出现,表明处方中其他共存成分对含量测定结果不存在相互干扰。
2.3.6 线性关系考察[12]分别精密移取“2.3.1”项下备用储备液0.125 mL、0.25 mL、0.5 mL、1.0 mL、2.0 mL置于10 mL 容量瓶中,加甲醇稀释至刻度,摇匀,分别精密吸取 20 μL,按“2.3.4”项下色谱条件分析,分别进样,记录色谱图,并进行积分处理。以色谱峰面积积分值(Y)为纵坐标,对照品溶液的质量浓度(X,μg/mL)为横坐标线性回归,得到鞣花酸的回归方程为Y=3.08×105X-1.09×105(r=0.9996);鞣花酸在质量浓度 0.57~9.05 μg/mL 内与峰面积呈良好的线性关系。
2.3.7 精密度考察 分别吸取浓度为 45.25 μg/mL 的鞣花酸对照品溶液10 μL,按“2.3.4”项下条件重复测定6次,以色谱峰面积积分值计算相对标准偏差(RSD),结果鞣花酸峰面积的RSD(n=6)为1.60%,表明仪器精密度良好。
2.3.8 重复性考察 分别精密称取六味石榴丸(批号:210802)样品细粉,6 份,按“2.3.2”项下方法制备供试品溶液,按上述色谱条件测定,得鞣花酸的平均含量和 RSD 分别为 79.41 μg/g、1.19%,表明该方法的重复性良好。
2.3.9 稳定性试验 取“2.3.2”项下同一供试品溶液,置于室温下,分别于0 h、2 h、4 h、8 h、12 h及24 h取样,进样分析,计算出鞣花酸峰面积积分值的RSD为 3.00%,提示供试品溶液在 24 h 内稳定。
2.3.10 加样回收率实验 取已知含量的六味石榴丸(批号:210802)样品细粉,精密称定,共6 份,每份 0.5 g,按样品含量100%分别加入鞣花酸对照品溶液,按“2.3.2”项下方法制备供试品溶液,依“2.3.4”项下色谱条件测定,并进行回收率的计算。测得鞣花酸的平均加样回收率为98.14%,RSD值为1.50%。
2.3.1.10 样品测定 精密称取来源于不同批号的各样品3份,每份1.0 g,按“2.3.2”项下方法制备样品溶液,依“2.3.1.4”项下色谱条件进样 20 μL,测定样品中鞣花酸的含量,结果见表2。
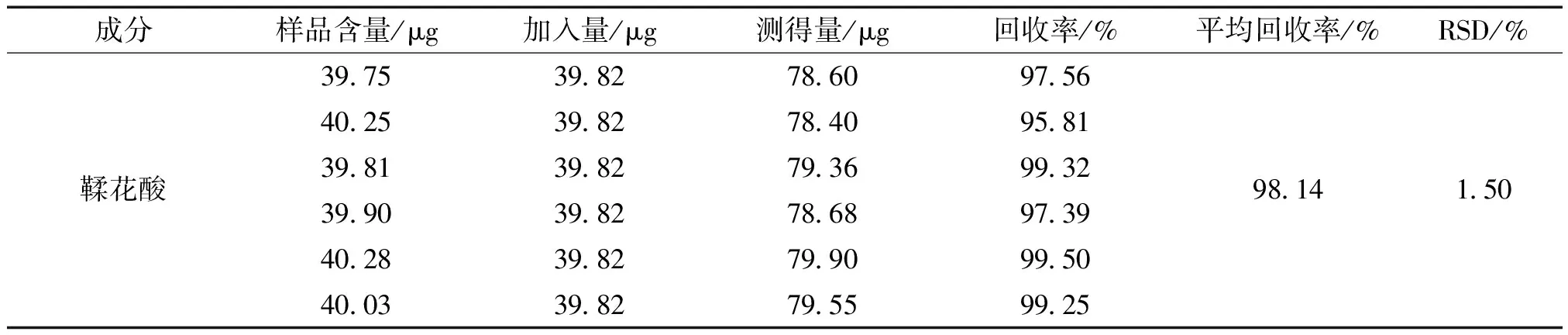
表1 加样回收率试验结果 (n=6)

表2 样品中鞣花酸含量测定结果 (n=3)
2.4 六味石榴丸中羟基红花黄色素A的HPLC含量测定
2.4.1 对照品溶液的配置 精密称取羟基红花黄色素A对照品 13.88 mg,置于25 mL容量瓶,用25%甲醇溶解、定容至刻线,摇匀,即得对照品储备液,备用。
2.4.2 供试品溶液的制备 取六味石榴丸适量研细,另取约0.3 g,精密称定,置具塞锥形瓶中,精密加入25%甲醇50 mL,称定重量,超声处理(功率300 W,频率40 kHz)50 min,放冷,再称定重量,用25%甲醇补足减失的重量,摇匀,静置后,取上清液经0.22 μm微孔滤膜,取续滤液作为供试品溶液。
2.4.3 阴性对照样品溶液的制备 按处方配比及制备方法制成不含红花药材的阴性对照样品,按“2.4.2”项下方法制备阴性对照样品溶液。
2.4.4 色谱条件 CAPCELL PAK-C18(250 mm×4.6 mm,5 μm);柱温:25 ℃;以甲醇-乙腈-0.7%磷酸溶液(26∶2∶72)为流动相;流速:1.0 mL/min;检测波长设置为403 nm。理论板数按羟基红花黄色素A峰计算,应不得低于4000。
2.4.5 专属性试验 分别上述对照品溶液、供试品溶液和阴性样品溶液,各10 μL,按“2.4.4”项下色谱条件检测。由图7可见,其分离度良好;阴性样品色谱图中,羟基红花黄色素A对应目标化合物在相应的保留时间无色谱峰吸收出现,表明处方中其他共存成分对含量测定结果不存在相互干扰。
2.4.6 线性关系考察 分别精密移取“2.4.1”项下备用储备液0.0626 mL、0.125 mL、0.25 mL、0.5 mL、1.0 mL、2.0 mL置 10 mL容量瓶中,加25%甲醇溶液定容至刻线,摇匀,分别精密吸取10 μL,按“2.4.4”项下色谱条件检测,分别进样,记录各色谱图。以色谱峰面积积分值(Y)为纵坐标,对照品溶液的质量浓度(X,μg/mL)为横坐标线性回归,结果羟基红花黄色素A的回归方程为Y=3.05×104X-4.91×104(r=0.9999);羟基红花黄色素A在质量浓度 6.72~107.50 μg/mL内与峰面积呈良好的线性关系。
2.4.7 精密度考察 分别精密吸取浓度为 53.75 μg/mL 的羟基红花黄色素A对照品溶液 10 μL,按“2.4.4”项下色谱条件重复测定6次,以色谱峰面积积分值计算其RSD值,结果羟基红花黄色素A峰面积的RSD(n=6)为0.67%,表明仪器精密度良好。
2.4.8 重复性考察 分别精密称取六味石榴丸(批号:210802)样品细粉,6 份,按“2.4.2”项下方法制备供试品溶液,按上述色谱条件进样分析,测得羟基红花黄色素A的平均含量和 RSD值分别为5.30 mg/g、1.99%,表明该方法的重复性良好。
2.4.9 稳定性试验 取“2.4.2”项下同一供试品溶液,室温下放置,分别于0 h、2 h、4 h、8 h、12 h、24 h取样,进样测定,计算得羟基红花黄色素A峰积分面积值的RSD 为0.84%,表明供试品溶液在24 h内稳定。
2.4.10 加样回收率实验 取已知含量的六味石榴丸(批号:210802)样品细粉,精密称定,供 6 份,每份 0.15 g,按样品含量100%分别加入羟基红花黄色素A对照品溶液,按“2.4.2”项下方法制备供试品溶液,依“2.4.4”项下条件,分析测定,并计算回收率。结果羟基红花黄色素A的平均加样回收率为99.00%,RSD值为1.34%。
2.4.11 样品测定 精密称取来源于不同批号的各样品3份,每份0.3 g,按“2.4.2”项下方法制备供试品溶液,依“2.4.4”项下色谱条件分析,并测定样品中羟基红花黄色素A的含量,结果见表4。

表4 样品中羟基红花黄色素A含量测定结果 (n=3)
3 讨论
3.1 显微及TLC定性鉴别 根据文献研究[3-4],选择处方药材粉末中典型显微特征,如花粉粒的形态、石细胞的特征等作为鉴别要点,可快速对处方投料情况进行甄别。同时,建立石榴子、肉豆蔻、胡椒及红花的TLC鉴别方法,各薄层色谱色谱条带清晰、分离度良好。另外,在藏区走访调研中,发现存在误将花椒代替胡椒的情况,已建立的TLC方法可进行有效鉴别。
3.2 HPLC测定 该制剂投料中石榴子占处方组成的28.6%,其主要特征成分为鞣花酸等鞣质类物质,具有显著的抗氧化、抗炎等活性[7],故选择鞣花酸作为指标性成分,对石榴子的质量状况进行考察。将鞣花酸对照品溶液,进行全波长扫描,确定252 nm为最佳检测波长。实验考察了乙腈-0.2%磷酸溶液(16∶84)、乙腈-0.2%磷酸溶液(18∶82)、乙腈-0.2%磷酸溶液(10∶90)等流动相体系,结果表明,以乙腈-0.2%磷酸溶液(16∶84)为洗脱系统,分离效果良好。通过对不同色谱柱温(25 ℃、30 ℃、35 ℃)的比较,30 ℃ 时,目标峰峰形良好,基线平稳。分别对提取溶媒(25%甲醇、50%甲醇、70%甲醇、甲醇)、提取方式(热回流、超声提取)及提取时间(10 min、20 min、30 min、40 min、50 min、60 min)进行考察,经综合考量最终选择以甲醇为提取溶剂,超声提取50 min。
六味石榴丸中红花,具有活血通经、散瘀止痛的功效,其中羟基红花素A是与功效相关的主要水溶性成分[13],且被《中国药典》作为红花的质控指标,其中所含羟基红花素A按干燥品计应不得少于1.0%,故选择羟基红花素A作为指标性成分,对红花投料的质量状况进行考察。对羟基红花素A对照品溶液,进行全波长扫描,确定 403 nm 为最佳检测波长。实验考察了甲醇-0.1%磷酸溶液(35∶65)、乙腈-0.1%磷酸溶液(35∶65)、甲醇-乙腈-0.7%磷酸溶液(26∶2∶72)等流动相体系,结果表明,以甲醇-乙腈-0.7%磷酸溶液(26∶2∶72)为最佳洗脱系统。通过对不同色谱柱温(25 ℃、30 ℃、35 ℃)的比较,选择25 ℃,目标峰峰形良好,基线平稳。分别对提取溶剂(10%甲醇、25%甲醇、50%甲醇、70%甲醇、甲醇)、提取方式(超声、热回流)及提取时间(20 min、30 min、40 min、60 min)进行考察,最终确定以25%甲醇为提取溶剂,超声提取 50 min,进行HPLC分析样品的制备。同时,按处方中红花投料比折算,六味石榴丸中羟基红花素A应不少于 1.4 mg/g。
综上所述,本研究从显微鉴别、TLC鉴别、含量测定等方面着手,较系统的建立了六味石榴丸的质量标准,操作方便便、专属性强、准确可靠,可为该制剂的质量控制提供参考。
猜你喜欢
中西医结合心脑血管病杂志(2022年12期)2022-07-07
快乐语文(2021年34期)2022-01-18
快乐语文(2021年27期)2021-11-24
音乐天地(音乐创作版)(2021年7期)2021-10-13
快乐语文(2021年11期)2021-07-20
草原歌声(2021年1期)2021-07-16
快乐语文(2021年15期)2021-06-15
少先队活动(2021年1期)2021-03-29
小天使·一年级语数英综合(2018年4期)2018-06-22
中成药(2017年4期)2017-05-17
