
长期培肥降低稻田土壤硝化和反硝化细菌功能基因丰度并减缓氮素周转
2023-10-24 09:46陈会巧马慧霞孙丽丽周昌敏顾文杰卢钰升徐培智解开治
植物营养与肥料学报 2023年9期
陈会巧,马慧霞,张 桥,孙丽丽,周昌敏,顾文杰,4,卢钰升,徐培智*,解开治*
(1 甘肃农业大学资源与环境学院,甘肃兰州 730070;2 广东省农业科学院农业资源与环境研究所 / 农业农村部南方植物营养与肥料重点实验室 / 广东省养分资源循环利用与耕地保育重点实验室 / 广东省土壤微生物与耕地保育工程技术研究中心,广东广州 510640;3 广东省农业环境与耕地质量保护中心,广东广州 510500;4 岭南现代农业科学与技术广东省实验室茂名分中心,广东茂名 525000)
稻田土壤由于其季节性淹水、干湿交替频繁发生而经常处于淹水还原、排水氧化的状态,导致硝化和反硝化作用发生极度活跃,进而影响稻田土壤氮循环过程[1]。其中,反硝化作用是微生物在厌氧条件下进行的硝酸盐还原过程,即NO3-→NO2-→NO→N2O→N2[2-4],在中间过程中产生温室气体N2O 的排放,反硝化作用一直受到广泛的关注[5]。亚硝酸盐(NO2-)还原为NO 的过程是第一步产气过程,也是反硝化过程的限速步骤[6-7],这一步骤由两种结构不同但功能相同的基因nirK和nirS编码的亚硝酸还原酶控制催化,因此nirS和nirK基因通常作为分子标记来研究环境中反硝化微生物的生物化学过程。研究表明,nirK和nirS基因的丰度与不同土壤环境因子有关[8],nirK和nirS型反硝化细菌群落结构也易受施肥方式的影响[9]。此外,反硝化作用通常伴随着硝化作用发生,硝化作用是将土壤中的氨转化为硝酸盐的过程,第一阶段是将氨氧化成亚硝酸盐的亚硝化过程,由氨单加氧酶和羟胺还原酶催化反应进行,氨单加氧酶将NH3氧化为NH2OH,再经hao催化氧化为NO2-;第二阶段是将亚硝酸氧化成硝酸盐的过程。其中AMO 由结构基因amoA、amoB和amoC构成,由于amoA具有序列保守性,而被作为AMO 功能的标记基因;hao则通过氧化羟胺为微生物的生长提供能量。功能标记基因的存在可以反映特定代谢过程的遗传潜力[10],硝化、反硝化功能基因的定量和表征也有助于将硝化和反硝化微生物类群与土壤氮循环潜力直接联系起来[11]。目前,对hao基因的研究主要是对其功能蛋白和基因结构的研究,集中于分离纯菌株[12-14],对它在土壤环境中的研究则较少。有研究以长期定位试验为平台,构建硝化基因克隆文库,研究长期施氮对氨氧化菌和亚硝化菌多样性及其群落结构的影响,结果表明,施氮肥使其多样性降低,群落结构也趋于单一化。土壤硝化与反硝化作用是土壤中决定植物利用的氮素有效性以及形态的关键过程,与温室气体释放和因硝酸盐淋失造成的水体污染引发的环境问题直接相关[15-17]。
多年来,考虑到稻田增值、高产的需要,化肥的施用已成为确保国家粮食安全和产量的关键措施。然而研究发现,长期不同施肥会引起稻田土壤理化性质出现差异,从而妨碍硝化和反硝化微生物的生长,再进一步影响稻田土壤硝化和反硝化作用[18-19]。研究表明,秸秆与肥料混合施用能显著增加土壤微生物数量,特别是固氮菌、硝化细菌、反硝化细菌和氨氧化细菌等氮转化功能细菌的数量,进而影响土壤氮循环。但也有研究表明,秸秆还田会导致土壤微生物多样性下降[20],对土壤微生物群落组成和功能也会有很大影响[21]。秸秆还田可维持表层土壤的淹水环境,并为土壤微生物提供大量的营养物质,土壤微生物可利用的营养物质会随着施入土壤中的秸秆不断分解而减少,也会进一步抑制土壤微生物活性[22]。土壤微生物的数量及活性主要取决于pH、有机质含量、土地利用类型等[23],当上述因素不断变化,且秸秆还田量和时间不同时,其对稻田微生物群落的影响也不尽相同[24]。
本研究依托位于江西红壤研究所长达40 年的长期定位试验平台,采用宏基因组测序和实时荧光定量PCR 技术,研究长期不同培肥处理对稻田土壤硝化和反硝化细菌群落及功能基因丰度的影响,旨在阐明硝化和反硝化功能微生物丰度及功能基因对不同培肥处理的响应及其关键控制因素,以期为稻田土壤养分管理和科学施肥提供基础数据支撑。
1 材料与方法
1.1 试验地概况
长期定位试验始于1981 年,地点位于江西省红壤研究所红壤生态站 (28°21′N,116°10′E)。该地区属于亚热带季风气候,夏热冬温,四季分明,海拔为25~30 m,晚稻季降水量占全年的14%;土壤类型为潴育型红壤水稻土[25]。初始耕层 (0—20 cm) 土壤理化性质如下[26]:pH 6.90、有机碳含量为16.30 g/kg、全氮含量为1.49 g/kg、全钾含量为10.40 g/kg、碱解氮含量为144.00 mg/kg、速效钾含量为80.50 mg/kg、有效磷含量为4.15 mg/kg、全磷含量为0.48 g/kg。
1.2 试验设计
本试验共设4 个处理,分别为:1) CK,不施肥;2) NPK,单施化肥;3) M1,早稻施绿肥紫云英22500 kg/hm2;4) M2,早稻施绿肥紫云英22500 kg/hm2+晚稻秸秆还田4500 kg/hm2。每个处理3 个重复,随机区组排列,每个试验小区面积64 m2,其他田间管理措施相同。试验水稻为当地主栽品种中早59,每5 年换1 次。绿肥品种为余叶大叶籽的紫云英 (AstragalussinicusL.),种植紫云英原地翻压,盛花期翻压量为22500 kg/hm2。紫云英和水稻秸秆的含水量分别为80.5%和49.2%,烘干后测定其养分含量,紫云英中N 含量为20.51 g/kg,K 含量为14.18 g/kg,P 含量为4.48 g/kg;水稻秸秆N 含量为10.80 g/kg,K 含量为18.60 g/kg,P 含量为2.37 g/kg。在施用有机肥的基础上,有机培肥处理 (M1 和M2) 每季额外施用化肥N 69.00 kg/hm2、P2O530.00 kg/hm2和K2O 67.50 kg/hm2。其中磷肥为钙镁磷肥 (P2O515%)、氮肥为尿素 (N46%)、钾肥为氯化钾 (K2O60%),不同处理的养分投入总量如表1 所示。

表1 长期培肥定位试验各处理养分投入量(kg/hm2)Table 1 Nutrient input of each treatment in the long-term experiment
1.3 样品采集与测定方法
1.3.1 样品采集 土壤样品采集于2020 年晚稻收获后。在各处理小区内,用土钻按五点采样法等量采集耕层土壤(0—20 cm)均匀混合,并采用四分法收集1 个土壤样品[27]。采集好的土壤样品立即置于带有冰袋的保温箱内,带回实验室在超级工作台中进一步处理。将土壤表层枝叶、植物根系等杂物去除,并充分混匀,然后分成两部分:一部分立即保存于-80℃冰箱中,用于分子生物学分析;另一部分自然风干后,用于土壤理化性质测定。
1.3.2 土壤理化性质测定 测定方法参考鲍士旦的《土壤农化分析》[28]。土壤pH 采用复合电极法 (水土体积比2.5∶1) 测定;全氮采用凯氏定氮消解法测定;碱解氮采用1 mol/L 的NaOH 碱解扩散法测定;硝态氮和铵态氮采用氯化钾浸提—流动注射分析法测定;有效磷采用氟化铵法测定;全磷采用HNO3-HF-HClO4消解—钼锑抗比色法测定;全钾采用火焰光度法测定;有机质采用重铬酸钾氧化外加热法测定;速效钾采用1 mol/L 乙酸铵浸提—火焰光度法测定;土壤微生物量碳氮的测定采用氯仿熏蒸浸提法。
1.4 土壤宏基因组测序及实时荧光定量PCR
称取0.5 g 储存于-80℃的土壤样品,使用FastDNA SPIN 试剂盒 (MP Bio-medicals,Solon,OH,USA) 提取土壤总DNA。采用NanoDrop 2000(Thermo Fisher,美国)测定提取液中DNA 的质量和浓度。检测合格的DNA 样品中加入fragmentation 缓冲液 (Thermo Fisher,美国),超声破碎仪随机打断后得到的短片段DNA 用于文库构建,质检合格的文库采用Illumina HiSeq 2500 高通量测序平台进行PE150 测序,每个样品测序数据量>10 G,对得到的宏基因组下机数据进行以下处理:数据质控→去宿主污染→序列组装→基因预测→基因序列聚类→物种功能注释。基于非冗余基因集的基因序列,使用BLASTP 软件与NCBI-NR 库进行物种同源性比对;还利用基于reads 的Metaphlan 2 软件对各样品的物种发布和组成进行分析,作为NR 分类法的补充。使用Diamond 软件将非冗余基因集Unigenes 与KEGG 的基因数据库进行基因同源性比对 (设定阈值≤0.001),为保证后续研究的生物学意义,从对比序列的结构中筛选出最低的e-value 作为该序列的对比结果。本研究中宏基因组数据经过预处理和质控后得到的Clean reads 共1.13×109条,进行聚类分析后得到的非冗余基因集Unigenes 平均长度为517.93 bp,平均GC 含量为62.19%,将非冗余基因集Unigenes 与KEGG 基因数据库进行比对注释,发现17891332 个非冗余基因集Unigenes 被注释到8301个基因。其中amoA有39 个Unigenes;hao有792个Unigenes;nirS有84 个Unigenes;nirK中有571个Unigenes。原始序列数据已存放在NCBI 序列,数据库注册编号为ID: PRJNA958469。
运用微生物碳氮磷硫元素循环功能基因的高通量定量技术,测定氮循环相关功能基因相对拷贝数,并以16S rRNA 作为内参对数据进行标准化得到各基因在各样本中相对定量,再根据Roche 仪器检测获得16S rRNA 基因的绝对定量,换算得到各基因的绝对定量信息。宏基因组测序及荧光定量PCR 工作委托广东美格基因科技有限公司完成,同时完成文库构建。
1.5 数据处理
采用Excel 软件进行数据整理,利用SPSS V22.0(IBM,USA)进行Shapiro-Wilk 正态性检验,来评估各处理理化性质、微生物功能基因数据的正态性分布,服从正态分布的数据使用基于post-hoc Tukey检验的单因素方差分析 (one-way ANOVA) 计算差异显著性;对不同类型微生物不同物种水平的物种组成进行统计后,使用Origin 2022 软件进行物种组成堆叠图的绘制;不同微生物和土壤理化特性的冗余分析 (redundancy analysis, RDA) 采用Canoco 5.0软件完成;ANOSIM 分析、NMDS 分析、Adonis检验、Spearman 相关分析采用R 语言中“vegan”和“pheatmap”软件包完成并绘图。
2 结果与分析
2.1 长期培肥对稻田土壤硝化和反硝化细菌群落多样性的影响
α 多样性指数(表2,表3) 表明,长期不同施肥处理对土壤硝化和反硝化细菌群落多样性和丰富度无显著影响。为揭示不同处理下的土壤硝化和反硝化细菌群落组成的差异性和相似性,通过相似性分析 (ANOSIM) 研究细菌群落组成的异同,并采用基于Bray-Curtis 距离的非度量多维尺度图 (NMDS) 展示(图1)。结果发现,各处理土壤硝化和反硝化细菌群落β 多样性与不施肥土壤样本点之间的距离都较大,进一步采用基于距离算法的Adonis 检验发现,长期不同培肥处理土壤硝化基因AOB (amoA) 细菌和反硝化基因nirK和nirS型细菌群落构成存在显著差异 (P<0.05),而基因hao细菌群落构成差异不显著。
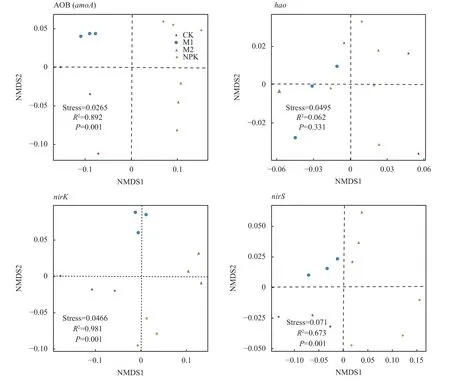
图1 不同处理土壤硝化和反硝化细菌NMDS 分析Fig.1 NMDS analysis of soil nitrifying and denitrifying bacteria in different treatments
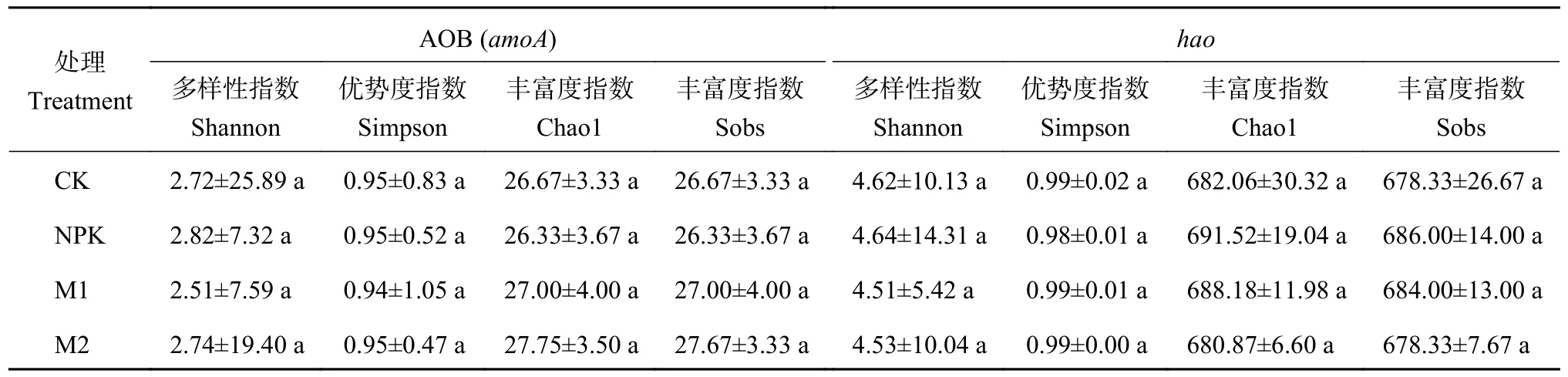
表2 长期培肥处理对土壤硝化细菌α 多样性的影响Table 2 Effects of long-term fertilization treatments on α diversity of soil nitrifying bacteria
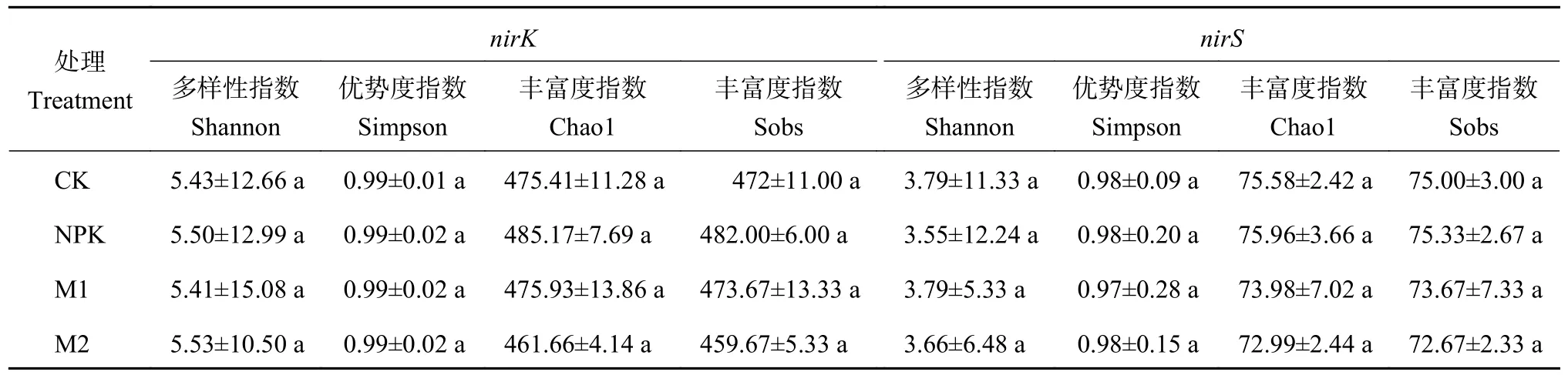
表3 长期培肥处理对土壤反硝化细菌α 多样性的影响Table 3 Effects of long-term fertilization treatments on α diversity of soil denitrifying bacteria
2.2 长期培肥对稻田土壤硝化和反硝化细菌群落组成的影响
通过序列比对分析作出相应硝化和反硝化功能基因的相对丰度图(图2),土壤硝化和反硝化菌群中含有大量分类地位不明确的微生物,按门水平划分各处理中nirS和nirK反硝化细菌以变形菌门(Proteobacteria) 占据着主导地位,AOB (amoA)和hao细菌同样以变形菌门(Proteobacteria) 为主导。按属水平分类选取丰度前十的菌属(图2),属水平下,已注释的氨氧化细菌AOB (amoA) 共有6 属,在CK和M1 处理中慢生根瘤菌 (Bradyrhizobium) 占比最高,分别为30%和32%,NPK 处理中的Methylocaldum菌属相对丰度占比高达30.6%,显著高于其他处理。甲基单胞菌属 (Methylomonas) 相对丰度表现为CK<NPK<M1<M2,在M2 处理中占比最大。硝化螺菌属(Nitrospira) 在CK 处理中的占比显著高于其他处理。

图2 不同处理硝化和反硝化细菌群落基于属分类水平的柱状堆叠图Fig.2 Columnar stacking diagram of nitrifying and denitrifying bacterial communities of each treatment based on genus classification level
已注释的hao细菌共有19 属,地杆菌属 (Geobacter) 相对丰度在各处理中占比最高。M2 处理中CandidatusKryptonium菌属相对丰度显著高于其他处理,硫磺菌属 (Sulfurifustis)在NPK 处理中相对丰度高于其他各处理,在NPK 和M1 处理中Stigmatella菌属的相对丰度高于CK 和M2 处理。地单胞菌属(Geomonas) 在各处理间无显著差异。
nirK反硝化细菌优势属主要有Ardenticatena菌属,占比最高,CK 处理为26.4% 和M1 处理为25.7%;布氏念珠菌 (CandidatusBrocadia) 菌属在CK 处理中占比最高,M2 处理中占比最少;硝化螺菌属(Nitrospira) 在NPK 处理和M2 处理中占比最高,分别达到17.1%和19.5%。罗河杆菌属(Rhodanobacter)相对丰度M2 处理显著高于其他处理(P<0.05)。
已注释的nirS反硝化细菌共有8 属,在NPK、M2 处理中类固醇杆菌属 (Steroidobacter)占比最高,分别为28% 和33%,NPK 和M1 处理中慢生根瘤菌属 (Bradyrhizobium) 占比最高,分别达到29%、24%。氢单胞 (Azohydromonas) 菌属相对丰度M1 处理显著高于其他处理(P<0.05),在M2 处理中相对丰度最低。
2.3 长期培肥土壤硝化和反硝化细菌群落结构与土壤理化性质的关系
基于RDA 分析,进一步揭示影响土壤硝化和反硝化细菌群落结构的主要环境因子,以土壤硝化和反硝化细菌属水平群落丰度为响应变量,土壤环境因子为解释变量(图3)。结果表明,nirK反硝化细菌群落在两个排序轴上的解释度分别为64.48% 和19.00%,Monte Carlo 检验表明,AOB (amoA)细菌群落在排序轴上的解释度分别为84.48%和6.24%,土壤速效氮 (P=0.004)、有效磷 (P=0.004) 和全磷(P=0.002) 对氨氧化细菌AOB (amoA) 群落结构产生显著影响。hao细菌群落在排序轴上的解释度分别为60.76%和26.80%,检验表明,土壤有机碳 (P=0.008)、有效磷 (P=0.01) 和速效钾 (P=0.008) 对hao细菌群落有显著影响。
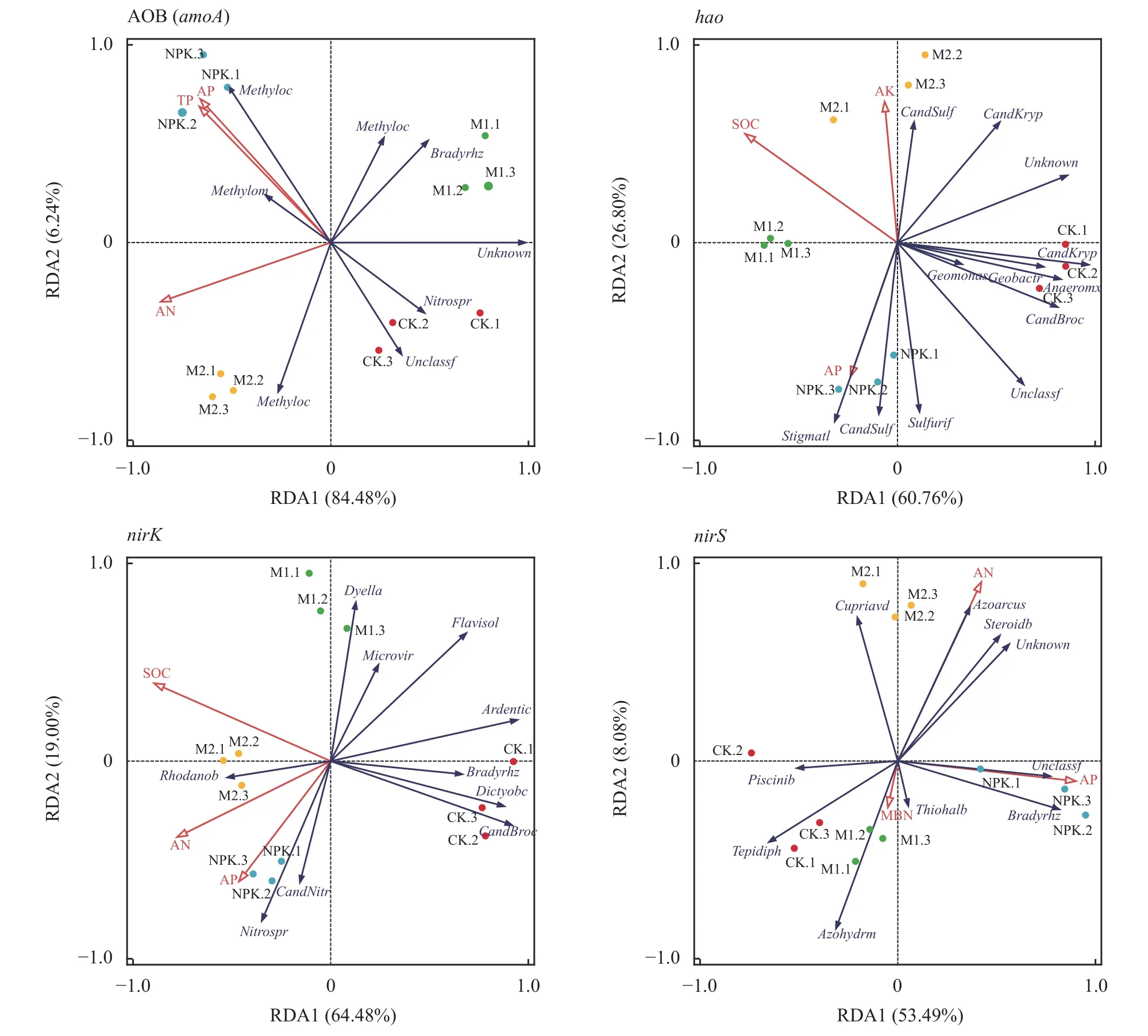
图3 不同处理土壤硝化和反硝化细菌与土壤因子RDA 分析Fig.3 RDA analysis of soil nitrification and denitrification bacteria and soil factors in different treatments
土壤速效氮 (P=0.002)、有效磷 (P=0.006) 和有机碳 (P=0.002) 对nirK反硝化细菌群落结构产生显著影响;nirS反硝化细菌群落在两个排序轴上的解释度分别为53.49%和8.08%,土壤有效磷 (P=0.022)对nirS反硝化细菌群落构成产生显著影响。
2.4 长期培肥对稻田土壤硝化和反硝化功能基因的影响
如图4 所示,硝化作用中amoA基因拷贝数明显高于hao基因,其中amoA基因拷贝数变幅为3.62×103~5.96×104copies/g,hao基因拷贝数为1.8×103~8.6×103copies/g,二者在不同处理下都逐渐下降,amoA基因拷贝数表现为CK 处理显著高于其他处理(P<0.05)。反硝化作用中编码含红血素cdl 的亚硝酸还原酶 (cd1-NIR)nirS基因拷贝数明显高于编码含Cu2+的亚硝酸还原酶 (Cu-NIR) 的nirK基因,其中nirK基因拷贝数变幅为7.8×103~4.2×104copies/g,nirS基因拷贝数为1.14×105~5.34×105copies/g。nirK、nirS基因拷贝数各处理与CK 处理相比时,不同培肥处理下都呈现下降趋势,nirK基因CK 处理显著高于其他处理 (P<0.05),nirS基因拷贝数在CK 和NPK 处理显著高于其他处理。
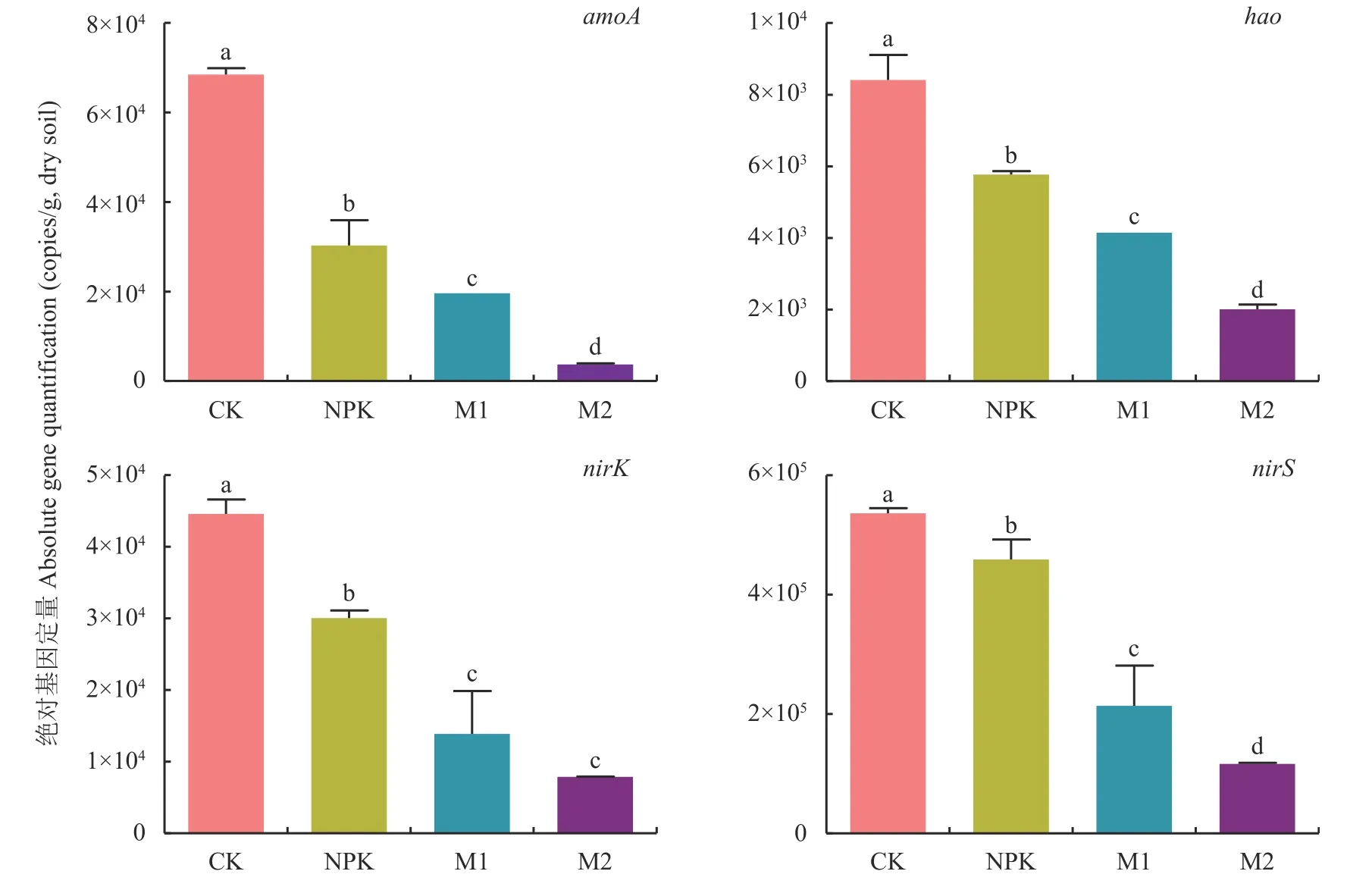
图4 不同处理土壤硝化和反硝化功能基因丰度Fig.4 Abundance of functional genes for nitrification and denitrification in soil under different treatments
通过Spearman 相关性分析(图5)表明,土壤有机碳和全氮与硝化功能基因(amoA和hao)、反硝化功能基因(nirK和nirS) 的丰度都呈极显著(P<0.01)负相关关系,硝化基因amoA丰度与速效氮、pH 和速效钾呈显著(P<0.05)负相关,hao基因与土壤速效氮、pH 和铵态氮为显著(P<0.05) 负相关。nirK反硝化基因丰度变化与土壤速效钾和铵态氮为显著(P<0.05)负相关,而nirS反硝化基因丰度与土壤pH、速效钾和铵态氮呈显著(P<0.05)负相关关系。
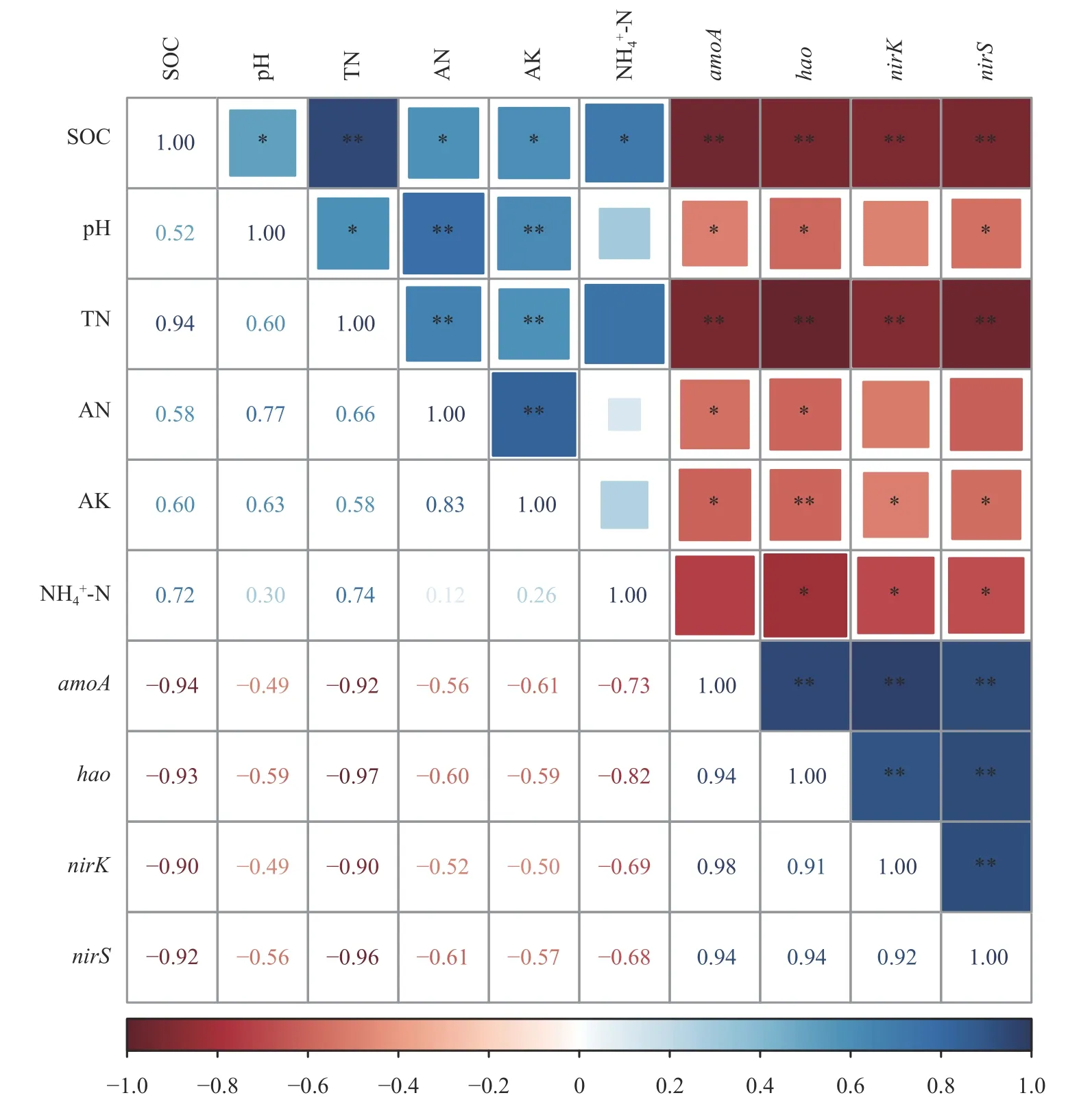
图5 土壤理化性质及功能基因丰度的Spearman 相关性分析Fig.5 Spearman correlation analysis on soil physical and chemical properties and functional gene abundance
3 讨论
3.1 长期培肥对稻田土壤硝化和反硝化细菌群落多样性及组成的影响
土壤细菌α 多样性指数是表征土壤生态系统稳定和细菌群落多样性的重要指标。有研究结果表明,pH 及土壤养分条件会影响土壤微生物α 多样性及丰富度,在一定范围内,pH、有机碳和全氮等含量差异越显著,土壤细菌的多样性差异越显著[29-30],本研究中,各培肥处理土壤pH、有机碳、全氮和有效磷等指标均高于CK 处理,而各培肥处理对硝化和反硝化细菌的α 多样性也均有提升,但不存在显著差异。Beta 多样性是研究物种群落间相似性程度的指数,在本研究中稻田土壤硝化和反硝化细菌的群落结构 (ANOSIM 结果有显著差异) 受长期培肥的影响较大,显著改变了nirK、nirS型反硝化细菌和氨氧化细菌AOB (amoA) 的结构组成,这可能是因为添加的有机物料作为土壤微生物的碳氮养分源,同时秸秆和紫云英自身也携带大量活的微生物,创建出一个富含营养的微生态环境,刺激了微生物的增殖,从而改变了土壤微生物群落组成和丰度。
不同施肥处理均会影响土壤细菌群落结构,总体上,硝化和反硝化细菌在各处理间的优势菌门相似,但在细菌属水平的相对丰度存在一定差异。不同施肥处理硝化和反硝化优势菌门均为变形菌门(Proteobacteria) 和酸杆菌门 (Acidobacteria),这与杨叶华等[31]研究得到的水稻土细菌群落优势类群相似。其中酸杆菌门是一种广泛分布在养分贫瘠和退化土壤中的寡营养型物种,其相对丰度与土壤有机质含量呈显著负相关[32]。本次研究中所检测到的硝化和反硝化细菌中未明确分类的相对丰度较高,已分类的硝化和反硝化细菌属水平相对丰度结果显示,nirS型反硝化细菌和AOB (amoA) 细菌中慢生根瘤菌(Bradyrhizobium)占比最高。AOB (amoA) 细菌中慢生根瘤菌 (Bradyrhizobium) 在各处理中占比最多,刘时光等[8]研究表明慢生根瘤菌 (Bradyrhizobium) 在一定程度上促进了农田土壤的硝化作用。与amoA基因情况不同,hao细菌群落多样性几乎未受到不同处理的影响,目前对于hao基因的研究还集中于分离纯菌株[33],对复杂土壤生态环境中的hao变化还无法进行解释。nirK型反硝化细菌的各菌属丰度中硝化螺菌属 (Nitrospira) 和罗河杆菌属 (Rhodanobacter) 在M2 (早稻紫云英+晚稻秸秆还田) 处理中丰度较高,可能由于该处理的施肥模式有利于有益菌属的增加。衷炜华等[34]研究也表明,罗河杆菌为好氧细菌,所以罗河杆菌属Rhodanobacter在此施肥模式中相对丰度较高。
3.2 长期培肥对稻田土壤硝化和反硝化细菌群落结构的影响
本研究结果表明,土壤碱解氮、有机碳、pH 和有效磷的差异是导致土壤硝化和反硝化细菌群落组成差异的重要因子。已有研究报道,土壤pH 对硝化和反硝化因子的影响最大[35],这可能是因为硝化作用和反硝化作用强烈依赖于土壤pH,而pH 的变化不仅制约着土壤养分的有效性而且能引起微生物在丰度上的响应[36]。长期施用化肥会引起土壤硝化作用及NO3-的淋溶进而导致pH 降低,土壤酸化严重,早稻施绿肥紫云英+晚稻秸秆还田处理提升了土壤pH,能够有效缓解土壤酸化[37]。早稻施绿肥的M1 处理微生物生物量氮含量最高,原因是豆科植物紫云英中氮素含量较高,可为微生物生长提供氮源,提高了微生物生物量,这与高嵩涓等[38]的研究结果相一致,其他各处理间微生物生物量氮含量无显著差异。pH 和土壤有机碳等土壤特性的变化会对参与土壤氮循环的功能群落产生影响[39-40],在长期施肥试验中,Chen 等[41]发现土壤总有机碳是控制反硝化菌群落大小变化的主导因素,但施肥仅对nirK型种群有显著影响,对nirS型种群没有显著影响。这与我们观察到的nirS和nirK型群落对施肥的响应一致。因此,施肥对nirS或nirK型群落结构的影响可能在很大程度上取决于土壤类型。陈娜等[42]研究发现,土壤有机碳和全氮是影响水稻土narG、nirK和nirS型反硝化细菌垂直分布的关键因子,而pH 是调控反硝化细菌在稻田底土中分布的核心驱动因子。Li 等[43]发现,不同功能群的相关基因对施肥处理的敏感性不同,其中nifH、napA和amoA基因对有机肥处理下的群落变异有显著影响。
3.3 长期培肥对稻田土壤硝化和反硝化功能基因的影响
由土壤硝化和反硝化功能基因丰度与土壤理化性质Spearman 相关性分析可知,影响稻田土壤硝化和反硝化功能基因丰度的主要土壤理化性质为土壤有机碳、全氮和NH4+-N,单施化肥模式amoA及hao功能基因丰度和反硝化过程nirK及nirS基因丰度均高于紫云英/秸秆还田模式。可能是因为秸秆的碳氮比较高,可增加微生物对无机氮的固持,使其转化为微生物量氮储存,进一步将有机氮的矿化作用降低。本研究中,4 个处理C/N 依次是7.69、8.23、8.51、8.28,相较于早稻施紫云英处理 (M1),早稻施紫云英+晚稻秸秆还田处理 (M2) 的土壤C/N 呈降低的趋势,因为豆科绿肥紫云英C/N 低,通过与根瘤菌共生固氮大幅提高氮素的积累,而禾本科水稻秸秆施入后由于分解缓慢导致养分固持。对于amoA基因,早稻紫云英和晚稻秸秆还田处理 (M2) 丰度较低,可能是因为富碳贫氮秸秆所引发的无机氮源被大量利用,因为秸秆添加提高了土壤有氧环境和碳含量,进一步刺激了有机氮的分解,产生了更多硝化作用的氮底物[44]。尽管增强了有机氮的分解,但在作物和其他微生物利用后,可供氨氧化微生物利用的氮可能不足,有机物料的添加可能使异养微生物快速增殖,压缩了氨氧化细菌和古菌的生存空间。对于反硝化功能基因来说,与CK 和NPK 相比,绿肥紫云英和秸秆还田则降低其基因丰度。这可能是由于有机物料碳氮比高于土壤,且腐解过程中形成微厌氧环境,还田后促进了硝酸盐异化还原成铵的过程,与反硝化微生物竞争底物,从而降低了反硝化功能基因的丰度[27]。马龙等[45]研究发现有机肥或秸秆替代部分化肥可显著降低土壤功能基因 (amoA和hao) 的丰度,且与土壤pH、全氮、有机质和铵态氮呈负相关。土壤氮含量和有机质含量在调节土壤氮循环方面发挥着举足轻重的作用,本研究结果表明,硝化和反硝化功能基因丰度均与铵态氮、全氮及有机碳含量呈显著负相关,故紫云英/秸秆还田模式可能通过影响土壤铵态氮及有机碳的含量,进而影响功能基因的表达。有研究发现[46-47]土壤硝化作用和反硝化基因nirK分别与NH4+-N 含量呈显著负相关,这与本研究的结果相一致。
4 结论
长期培肥显著影响了稻田土壤硝化和反硝化细菌群落的β 多样性,改变了nirK、nirS型反硝化细菌和AOB (amoA)细菌的群落组成。长期培肥处理中,绿肥紫云英和秸秆还田处理对稻田土壤硝化和反硝化细菌群落结构提升更显著,有效磷、速效氮和有机碳含量是影响nirS、nirK型反硝化细菌和AOB (amoA)、hao细菌群落结构的主要环境因子。
长期培肥显著降低了稻田土壤amoA基因和nirK、nirS型反硝化基因的丰度。土壤有机碳和全氮含量与硝化过程功能基因 (amoA和hao)、反硝化过程功能基因 (nirK和nirS) 的丰度都呈现极显著(P<0.01) 负相关关系。就硝化、反硝化功能基因丰度而言,长期培肥抑制了稻田土壤氨氧化过程及反硝化作用,延缓了稻田土壤氮循环的周转。
猜你喜欢
趣味(作文与阅读)(2021年5期)2021-08-19
作文大王·低年级(2019年2期)2019-01-23
创新作文(小学版)(2018年19期)2018-11-30
中学时代(2018年6期)2018-11-20
启蒙(3-7岁)(2018年8期)2018-08-13
海峡姐妹(2018年7期)2018-07-27
环境保护与循环经济(2017年7期)2018-01-22
作文与考试·小学高年级版(2016年14期)2016-09-10
中国环境科学(2016年3期)2016-02-08
应用海洋学学报(2014年2期)2014-11-26
