
茶叶和土壤中cis-联苯菊酯、高效氯氟氰菊酯手性对映体残留分析
2023-10-23 08:14:22宁亚婷王新茹罗逢健李建勋张新忠
核农学报 2023年11期
宁亚婷 王新茹 罗逢健 李建勋 张新忠,
(1中国农业科学院茶叶研究所,浙江 杭州 310008;2中国农业科学院农产品加工研究所,北京 100193)
茶叶是我国出口竞争力较强的传统特色农产品。目前我国茶树种植面积和产量都居全球之首,茶叶出口居世界第二。茶树在生长过程中常遭遇茶尺蠖、茶假眼小绿叶蝉、粉虱和象甲等多种害虫危害,严重影响品质和产量。目前,占世界杀虫剂市场30%~40%的拟除虫菊酯类农药是防治茶园害虫的主要农药。例如,cis-联苯菊酯、高效氯氟氰菊酯等是我国无公害茶园防治假眼小绿叶蝉、茶尺蠖等的推荐首选品种,为保障茶叶产量和茶农创收做出了巨大贡献。但其在茶叶和环境土壤中的残留,也带来了茶叶质量安全和环境污染隐患。
1999 年,Lewis 等[1]在《Nature》上发表了环境因素对手性农药甲基-2,4-D丙酸降解的影响,提出环境中手性农药潜在生物效应如毒性、致癌性、致突变性、内分泌干扰性以及环境中的持久性等大都具有对映体选择性差异,引起了各国科学家的广泛关注。国内外在手性农药拆分[2-5],水生生物急性毒性[6-7],土壤[8-9]、水[10-11]及动物[12-14]、植物[15-17]体内选择性降解环境行为等方面的研究较多。目前市场上大多数拟除虫菊酯类农药多以外消旋体销售[18-19],其中无活性功能的对映体存在,不仅增加了农药的使用量,导致作物和食品中残留量加大,而且会对非靶标生物产生负面影响[20]。近年来,大量手性拟除虫菊酯类农药对映体在水生生物毒性选择性研究[21-23]表明,cis-联苯菊酯和高效氯氟氰菊酯均存在对映体毒性选择性差异。人体是由左旋氨基酸组成的生命体,不能很好地代谢右旋分子,因此摄入含有右旋分子的食品或药物会成为人体代谢的负担,甚至对生命造成危害。目前世界主要茶叶生产、消费国或地区对茶叶中cis-联苯菊酯、高效氯氟氰菊酯的残留量均设定了最大残留限量值(maximum residue limit,MRL),介于0.01~25 mg·kg-1之间[24],但仅对外消旋体的总量进行了限制,并未考虑不同对映体之间的差别。降解规律的研究往往也很少考虑对映体间的差异,如鲜叶上两种手性农药对映体之间的降解速度是否一致?归趋是否相同?截至目前,针对茶叶和土壤中cis-联苯菊酯和高效氯氟氰菊酯两种菊酯类手性农药对映体的同时检测方法及其在茶鲜叶中选择性降解行为研究仍鲜见报道。
本研究通过对色谱柱和提取条件进行优化,建立起不同茶叶、土壤中cis-联苯菊酯和高效氯氟氰菊酯农药手性对映体的残留分析方法,并对方法学进行全面评价,旨在为进一步研究两种农药对映体在茶叶生长—加工—饮用过程中的选择性降解行为提供精准可靠的技术手段。
1 材料与方法
1.1 仪器与试剂
Varian 3800 GC 气相色谱仪(配备电子捕获检测器、1077 进样口、8410 自动进样器和Version 6.9.1 Saturn工作站),美国Bruker公司;Sigma 3k-5高速离心机,德国Sigma 公司;DFT-200 手提式高速万能粉碎机,浙江温岭市林大机械有限公司;T18 高速均质匀浆仪,德国IKA公司;电子分析天平(0.000 1 g和0.01 g),瑞士梅特勒-托利多公司;R-210 旋转蒸发仪,瑞士BUCHI公司;KQ-250DB 型数控超声波清洗器,昆山市超声仪器有限公司;玻璃柱管(18 mm I.D.×200 mm L.),杭州常盛科教器具厂;0.22 μm Filter Unit 滤膜,天津博纳艾杰尔科技有限公司。
正己烷、丙酮(色谱纯),美国Honeywell 公司;NaCl、无水Na2SO4(分析纯),上海试四赫维化工有限公司;硅镁型吸附剂(Florlisil 填料,60~100 目,使用前650 ℃烘4 h,冷却后按10%比例加入纯净水密封振荡混匀4 h,放置干燥器中备用),温州市化学用料厂;Florisil 与GCB 混合柱:取5.0 g Florlisil 与0.10 g GCB混合均匀得混合物,按照1 g Na2SO4、Florisil+GCB 混合物、1 g Na2SO4,从下往上先后装入玻璃柱管中,10 mL正己烷+丙酮(95∶5,V/V)预淋洗。
cis-联苯菊酯和高效氯氟氰菊酯外消旋体标准品(R体∶S体=1∶1,98%),德国Dr.Ehrenstorfer公司。
1.2 色谱条件
色谱柱:Daicel ChiralPak®AS-H手性色谱柱(4.6 mm ID×250 mm×5 μm,日本Daicel 公司),Phenomenex®Cellulose-1 手性色谱柱和Phenomenex®Cellulose-3 手性色谱柱(4.6 mm ID ×250 mm×5 μm,美国Phenomenex公司),BGB-172 手性气相色谱柱(30 m×0.25 mm ID×0.25 μm,瑞士BGB Analytik AG 公司),Cyclosil-B 手性气相色谱柱(30 m×0.32 mm ID ×0.25 μm,美国安捷伦科技有限公司);载气为N2,流量2.0 mL·min-1;进样口温度:260 ℃;进样量:1 μL;进样方式:不分流进样;ECD 检测器温度:300 ℃,CAP 模式;尾吹气N2,流量50 mL·min-1;色谱柱升温程序:初始180 ℃,保持5 min;以5 ℃·min-1升至230 ℃,保持80 min。
1.3 样品处理
分别取粉碎后的茶叶(绿茶、红茶、普洱茶、茶鲜叶)和土壤样品,称取5.00 g,加入正己烷+丙酮(4+1,V/V)混合溶液50 mL浸泡过夜,15 000 r·min-1均质匀浆提取1 min,加入2.0 g NaCl,混匀超声10 min,过滤提取液转移至圆底烧瓶,并用30 mL 提取溶剂重复提取残渣1 次,合并2 次提取液,浓缩近干,5 mL 正己烷+丙酮(95+5,V/V)溶解上样至Florlisil+GCB 混合柱,继续用30 mL 正己烷+丙酮(95+5,V/V)洗脱接收,浓缩近干,环己烷+丙酮(70+30,V/V)超声溶解定容1.5 mL,进样1 μL,气相色谱-电子俘获检测器(gas chromatographyelectron capture detector,GC-ECD)测定,外标法定量。
1.4 标准曲线与检出限
分别称取0.010 0 gcis-联苯菊酯和高效氯氟氰菊酯外消旋体标准品至烧杯中,正己烷溶解并转移到50 mL 容量瓶定容,配制成200 mg·L-1(对映单体浓度为100 mg·L-1)标准储备液,-18 ℃下避光保存。将储备液用正己烷稀释成对映单体浓度为800、400、200、100、50、25、12.5、5.0、2.5 μg·L-1系列标准工作溶液,GC-ECD 进样1 μL,以浓度为横坐标x,对映体重复测定3 次色谱峰面积平均值为纵坐标Y,获得cis-联苯菊酯和高效氯氟氰菊酯两对对映体溶剂标准曲线、线性相关系数及仪器检出限。
1.5 添加回收率、精密度与方法定量限
分别称取经测定不含cis-联苯菊酯和高效氯氟氰菊酯残留的空白绿茶(绿茶、红茶、普洱茶、茶鲜叶)和土壤样品5.00 g,分别添加外消旋体浓度为0.050、0.50 和5.00 mg·L-1标准溶液1.00 mL,相当于对映单体0.005、0.05 和0.50 mg·kg-1的3 个添加浓度水平,每个添加浓度平行试验6 份,涡旋混匀后放置过夜以达到更接近于实际样品,另外各自进行4 份空白试验,按照1.3节进行提取净化,其中3份空白用于配制对应浓度的基质标准溶液,作为基质标准进行测定,计算添加回收率、相对标准偏差及方法定量限,并通过基质标准溶液与溶剂标准溶液响应对比基质效应(matrix effect,ME),计算公式如下:
式中,A代表在纯溶剂中对映单体的响应值;B代表基质标准溶液中对映单体的响应值。
2 结果与分析
2.1 手性色谱分离条件的选择优化
为了实现cis-联苯菊酯和高效氯氟氰菊酯两对对映体一次进样拆分并获得对映单体,本研究首先对比了正相液相色谱条件下,20 ℃时不同类型色谱柱(Daicel ChiralPak®AS-H、Phenomenex®Lux 5 μm Cellulose-1和Phenomenex®Lux 5 μm Cellulose-3 柱)、不同流量(0.20~1.00 mL·min-1)及不同比例正己烷和异丙醇(99∶1~80∶20,V/V)对cis-联苯菊酯和高效氯氟氰菊酯对映体的拆分效果。结果表明,Cellulose-1柱对cis-联苯菊酯没有拆分效果,对高效氯氟氰菊酯有一定的拆分效果,在1%异丙醇条件下即可拆分高效氯氟氰菊酯为两个峰。增加异丙醇的比例可以改善峰型、减小峰宽、缩短保留时间,但会降低分离度,在20%异丙醇时仅能够基本拆分高效氯氟氰菊酯。AS-H能拆分高效氯氟氰菊酯对映体,但对cis-联苯菊酯无拆分效果。Cellulose-3 对cis-联苯菊酯、高效氯氟氰菊酯对映体均有一定拆分效果,在20%异丙醇时实现对cis-联苯菊酯对映体的拆分,但无法拆分高效氯氟氰菊酯对映体;随着异丙醇比例和流动相流量的降低,高效氯氟氰菊酯对映体逐渐实现分离,cis-联苯菊酯和高效氯氟氰菊酯对映体保留时间(retention time,tR)延长、分离度(resolution,Rs)增加、色谱峰变宽(表1)。在一定流动相条件下,干扰峰(tR=4.92 min)会对cis-联苯菊酯-1(tR=5.32 min)造成干扰(图1)。最终选取Phenomenex®Lux 5 μm Cellulose-3柱作为分析柱,正己烷+异丙醇(95∶5,V/V)在0.80 mL·min-1流量下进行拆分,0.25 mg·L-1标准溶液拆分效果见图1,cis-联苯菊酯两个对映体tR分别为5.32和5.98 min,高效氯氟氰菊酯两个对映体tR分别为11.22 和12.39 min,对映体分数(enantiomer fractions,EFs)分别为0.513和0.493。但本试验对5 g茶叶样品进行净化后分析发现(图1),正相液相色谱紫外检测器法(normal high performance liquid chromatographyultra-violet,NHPLC-UV)虽然可以满足对映体拆分的需要,但是茶叶基质富含咖啡碱等在紫外下强响应的杂质,前处理无法完全除去,导致背景响应高,对cis-联苯菊酯和高效氯氟氰菊酯对映体掩盖干扰严重,从而无法满足残留分析的需要。
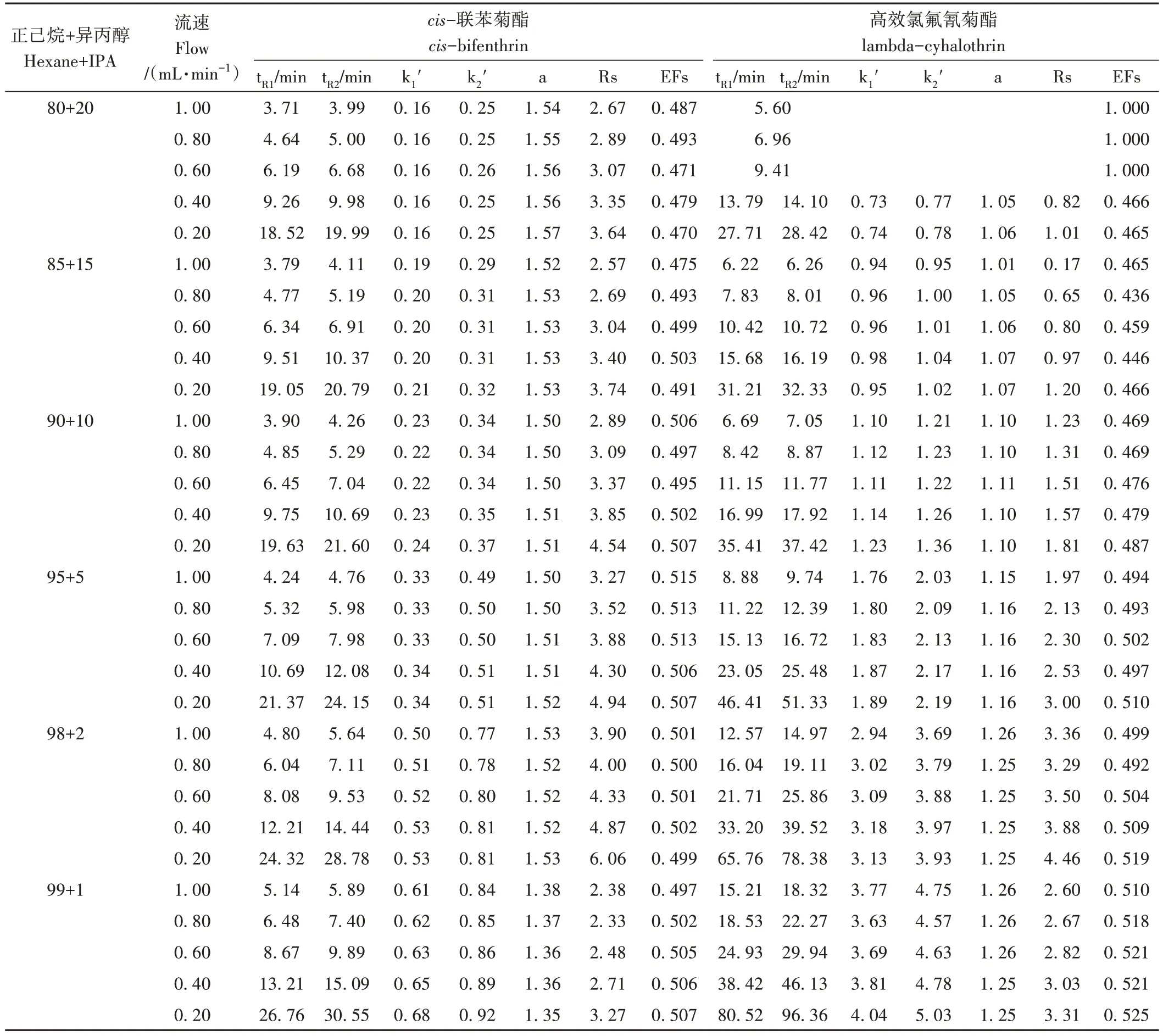
表1 不同条件下Cellulose-3柱对cis-联苯菊酯、高效氯氟氰菊酯对映体的拆分结果Table 1 The separation results for the enantiomers of cis-bifenthrin and lambda-cyhalothrin on Cellulose-3 column under different conditions
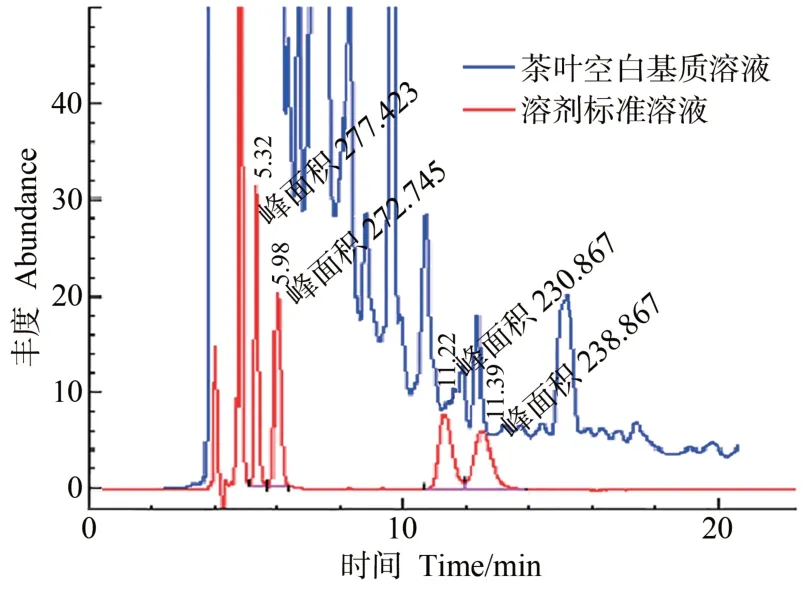
图1 净化后茶叶基质溶液与0.25 mg·L-1 cis-联苯菊酯和高效氯氟氰菊酯对映体标准溶液对比HPLC-UV色谱图Fig.1 The HPLC-UV chromatograms of purified tea matrix solution and 0.25 mg·L-1 solvent standard solution for enantiomers of cis-bifenthrin and lambda-cyhalothrin
ECD为破坏性检测器,无法获得对映体单体,但拟除虫菊酯类农药在GC-ECD 上有高灵敏度的响应,适合残留分析。有研究表明,BGB-172 GC柱能很好地分离cis-联苯菊酯手性对映体,并利用GC-ECD进行了环境水[25]、生物样本[22]中的对映体残留分析。在此基础上,本研究对比了BGB-172 GC 柱和Cyclosil-B GC 柱对cis-联苯菊酯和高效氯氟氰菊酯两组手性对映体拆分的效果,优化了色谱升温程序,对茶叶和土壤中cis-联苯菊酯、高效氯氟氰菊酯手性对映体进行了残留分析研究。结果表明,同样条件下,Cyclosil-B GC柱无法拆分对映体,BGB-172 GC柱在不同升温程序下有不同的分离效果,改变载气类型氦气(He)或氮气(N2),BGB-172 GC 柱下对两组手性对映体分离效果无明显差别,最终选择1.2 节色谱条件中色谱升温程序,能实现对映体的基本分离用于残留分析,色谱图见图2,此时cis-联苯菊酯对映体tR分别为42.88 和43.32 min,高效氯氟氰菊酯对映体tR分别为59.69 和60.45 min,既能实现cis-联苯菊酯和高效氯氟氰菊酯两组手性对映体的同时拆分,又能满足残留分析的灵敏度需要,同时相对于文献[26-27]缩短了对映体拆分时间。
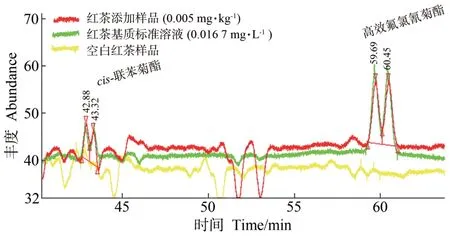
图2 空白红茶样品、0.005 mg·kg-1水平下红茶添加样品和0.016 7 mg·L-1红茶基质标准溶液GC-ECD色谱图Fig.2 The GC-ECD chromatograms of blank black tea,spiked black tea at 0.005 mg·kg-1,and black tea matrix standard solution sample at 0.016 7 mg·L-1 for cis-bifenthrin and lambda-cyhalothrin enantiomers
2.2 提取净化方法的优化
进行手性农药对映体残留分析时,既要尽量完全分离对映异构体,又要避免杂质干扰。当采用GCECD 分析茶叶样品时,由于ECD 是选择性的高灵敏度检测器,只对如含卤素、硫、磷、氮的电负性物质有信号,而对电中性(无电负性)的物质,如烷烃等无信号,因此,茶叶和土壤中未净化完全的大多数杂质在ECD上无明显信号或者能被分离开,从而不会像在液相紫外检测中那样造成背景干扰。
本试验用丙酮提取目标物,提取液浓缩近干后,采用5.0 g Florisil与0.10 g GCB混合柱净化,加入0.10 g的GCB 用来吸附除去部分色素,既能节省试验成本(相对于QuEChERs 等方法净化),又能更多地去除咖啡碱、儿茶素等杂质共提物,在保证净化效果的同时降低了后期ECD仪器的维护。由图3可知,正己烷+丙酮(95+5,V/V)洗脱时回收率和净化效果最佳。最终选择对5 g 茶叶或土壤样品加入正己烷+丙酮(4+1,V/V)混合溶液提取后,再利用5.0 g Florisil+0.10 g GCB 混合柱净化,30 mL 正己烷+丙酮(95+5,V/V)作为洗脱剂进行洗脱接收,浓缩近干后,定容,GC-ECD检测。
2.3 标准曲线、添加回收率、精密度和定量限
在1.2节色谱条件下,2.5~800 μg·L-1(2.5、5、12.5、25、50、100、200、400和800 μg·L-1)浓度范围内,cis-联苯菊酯对映体色谱峰面积(Y)与浓度(x)之间分别满足线性方程Y=15 381.1x-279 657.5(r=0.996 7)和Y=17 387.6x-399 048.0(r=0.993 7),对映体分数EFs=0.489±0.017;在2.5~200 μg·L-1(2.5、5、12.5、25、50、100和200 μg·L-1)浓度范围内,高效氯氟氰菊酯对映体色谱峰面积(Y)与浓度(x)之间分别满足线性方程Y=22 083.8x-161 004.3(r=0.992 3)和Y=23 001.1x-167 694.8(r=0.992 2),对映体分数EFs=0.492±0.008。
在0.005、0.050和0.50 mg·kg-1(低、中、高)3个添加浓度水平下,按照1.5 节进行试验,不同空白茶叶、土壤样品中添加回收率和精密度结果见表2。结果表明,cis-联苯菊酯、高效氯氟氰菊酯两对手性对映体在不同茶叶、土壤中的平均添加回收率(average spiked recovery,A.R.)为61.6%~123.4%,相对标准偏差(relative standard deviations,RSDs)为2.5%~14.6%。其中绿茶中回收率为63.0%~88.3%,RSDs为5.7%~13.8%;红茶中回收率为81.4%~121.7%,RSDs为2.5%~14.6%;普洱茶中回收率为61.6%~123.4%,RSDs为3.5%~11.7%;茶鲜叶中回收率为75.6%~102.6%,RSDs为4.6%~12.9%;土壤中回收率为68.3%~98.5%,RSDs为4.5%~14.2%。红茶空白、低浓度添加和基质标准溶液色谱图见图2,仪器检测限均低于1 μg·L-1,方法定量限均低于0.005 mg·kg-1。说明该方法能够满足茶叶和土壤中cis-联苯菊酯、高效氯氟氰菊酯残留检测的需要。通过3 个浓度下基质标准与溶剂标准响应对比,未见到明显的基质增强或减弱效应。因此,在实际测定时,可直接用溶剂标准进行定量,亦能确保定量准确度。
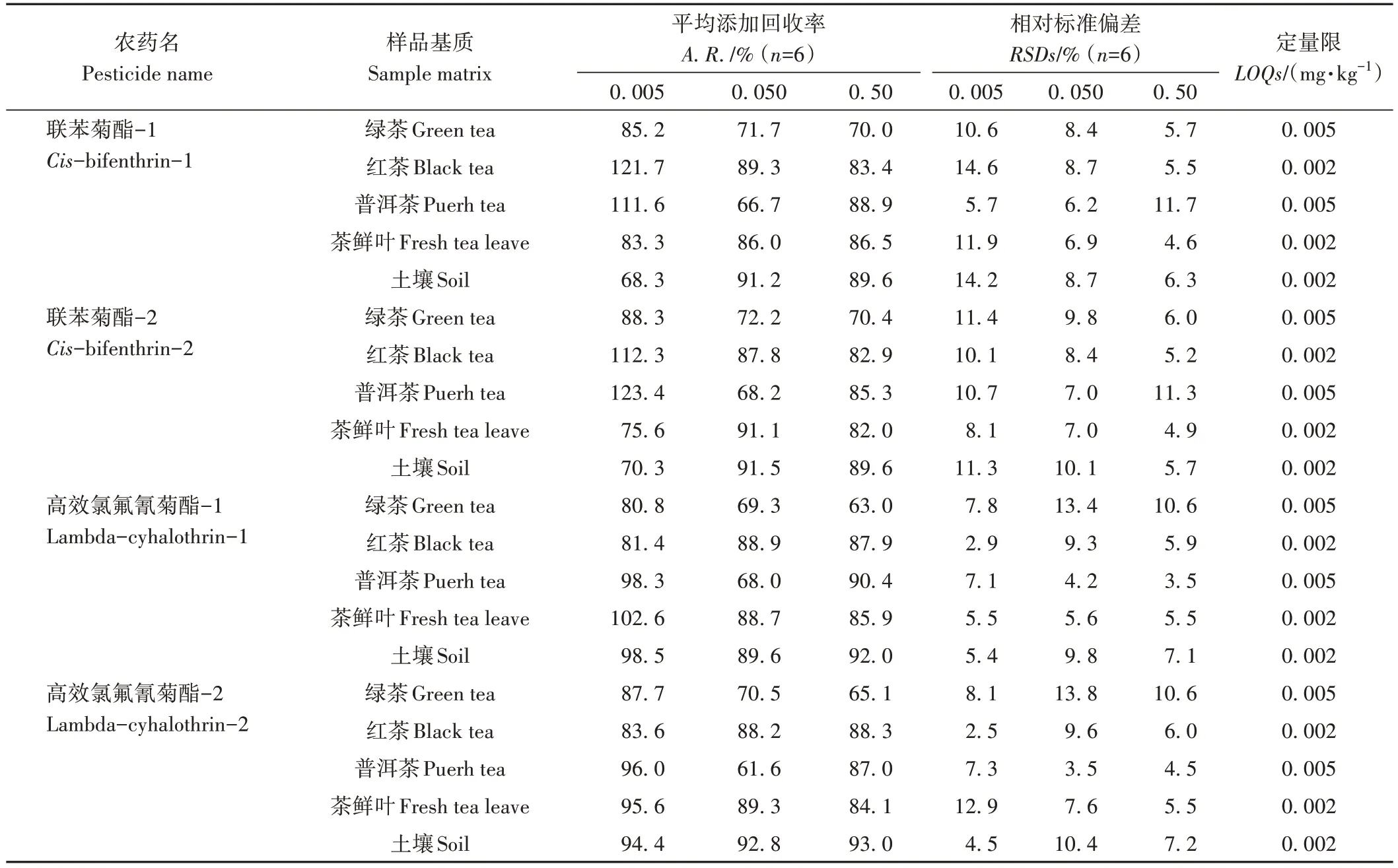
表2 GC-ECD测定不同茶叶和土壤样品中cis-联苯菊酯和高效氯氟氰菊酯对映体的平均添加回收率、相对标准偏差和定量限Table 2 The average spiked recoveries,relative standard deviations (RSDs) and limits of quantifications (LOQs) of cis-bifenthrin and lambda-cyhalothrin enantiomers in different tea,and soil samples by GC-ECD determination
2.4 两组对映体在茶鲜叶中的降解实际样品测定
利用建立的不同茶叶中手性农药cis-联苯菊酯和高效氯氟氰菊酯对映体残留GC-ECD 分析方法,研究了茶园鲜叶喷药cis-联苯菊酯和高效氯氟氰菊酯农药后,喷药后分别间隔2 h 和1、2、3、5、7、10、14、21 d 后采样,测定茶叶鲜叶中cis-联苯菊酯和高效氯氟氰菊酯两组手性对映异构体的残留量,获得其降解规律,结果表明,茶鲜叶中cis-联苯菊酯两个对映体残留量(Y,mg·kg-1)与时间(X,d)之间的关系分别符合曲线Y=4.857 3e-0.2048X,R2=0.911 9,半衰期t1/2=3.38 d和Y=5.153 5e-0.2012X,R2=0.897 4,半衰期t1/2=3.44 d;高效氯氟氰菊酯两个对映体残留量(Y,mg·kg-1)与时间(X,d)之间的关系分别符合曲线Y=0.409 8e-0.2166X,R2=0.964 2,半衰期t1/2=3.20 d 和Y=0.401 7e-0.2164X,R2=0.963 6,半衰期t1/2=3.20 d,见图4,通过半衰期可知,两种菊酯类农药对映体在茶鲜叶生长过程中的降解无明显的选择性差异。
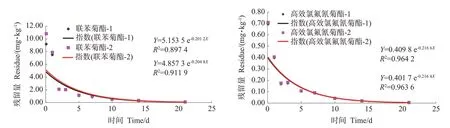
图4 茶鲜叶中cis-联苯菊酯和高效氯氟氰菊酯对映体降解曲线Fig.4 The enantioselective degradation curve of cis-bifenthrin and lambda-cyhalothrin in fresh tea leaves
3 讨论
目前针对拟除虫菊酯类农药对映异构体的分析通常是采用不同的色谱条件。Ye 等[3]系统总结了正相液相色谱条件下,单手性中心拟除虫菊酯类农药采用Chiralcel OD 柱和多手性中心拟除虫菊酯类农药采用串联Chirex 00G-3019-OD 柱拆分的效果;刘一平等[28]采用Chiralcel®OD-H 色谱柱拆分了高效氯氟氰菊酯对映体;陆娴婷等[26]采用Chiralcel OJ-H 柱拆分了cis-联苯菊酯对映体;文献[27,29]报道了采用手性气相色谱柱BGB-172 分离氯氰菊酯对映体,但目前还未见采用一根色谱柱同时对cis-联苯菊酯和高效氯氟氰菊酯对映体拆分的报道。本研究通过对比液相和气相色谱条件下不同色谱柱分离测定效果,优化分析条件,最终实现了cis-联苯菊酯和高效氯氟氰菊酯对映体的同时拆分。
茶叶基质复杂,富含咖啡碱、色素等杂质,因此,在对残留农药分析前进行提取、浓缩和净化十分重要。目前,茶叶中cis-联苯菊酯和高效氯氟氰菊酯残留测定中前处理技术主要是固相萃取法和QuEChERS 方法,如王艳丽等[30]采用固相萃取结合气相色谱-三重四极杆串联质谱法测定茶叶中45 种农药残留;许芮菡等[31]采用QuEChERS 方法,即乙腈提取后多壁碳纳米管净化,最后采用气相色谱-串联质谱分析了茶叶中的拟除虫菊酯类农药残留;国标GB 23200.113—2018[32]中也规定了茶叶中cis-联苯菊酯和高效氯氟氰菊酯残留分析的固相萃取法和QuEChERS前处理方法。相比固相萃取法,QuEChERS 方法较为简单,应用性也更广泛。本研究方法和许芮菡等[31]方法中测定cis-联苯菊酯和高效氯氟氰菊酯的定量限(均为0.005 mg·kg-1)均低于国标GB 23200.113—2018(0.005 mg·kg-1),表现出良好的检测灵敏度。而许芮菡等[31]采用改进的QuEChERS 方法提取净化,采用乙腈提取茶叶样品,乙腈会溶解并提取出各种极性与非极性目标物及杂质,杂质过多容易污染ECD 检测器,同时乙腈还会损坏气相色谱柱,需要转换进样溶剂为正己烷。而cis-联苯菊酯和高效氯氟氰菊酯是弱极性农药,本研究根据茶叶基质和待测成分理化特性,采用正己烷和丙酮混合溶剂作为提取溶剂,能够有效减少极性杂质的提取量,利用正己烷与待测成分之间的相似相溶原理及丙酮的强浸润性,从而保证了目标物的提取回收率和平行性,同时也降低了对ECD 检测器的污染。此外,本研究方法可以明确分析两种菊酯类农药对映单体的含量,而许芮菡等[31]的方法和国标GB 23200.113—2018 均无法实现。
cis-联苯菊酯和高效氯氟氰菊酯对映体在茶鲜叶生长过程中的降解无明显的选择性差异,主要是因为鲜叶基体作用下的对映体降解差异不显著。鲜叶采摘后的加工过程复杂,温度、湿度、pH、微生物等作用都可能导致对映体的选择性降解,这将是后续研究的重点,即通过系统研究两种菊酯类农药对映体在茶叶生长-加工-饮用过程中的选择性降解行为,为其更精准的风险评价提供基础数据和技术支撑。
4 结论
本研究首次对比分析了正相液相色谱条件下,Daicel ChiralPak®AS-H、Phenomenex®Cellulose-1 和Cellulose-3 柱对cis-联苯菊酯和高效氯氟氰菊酯两组手性对映体的拆分效果,以及利用气相色谱BGB-172柱同时分离测定效果,建立了茶叶和土壤中手性农药cis-联苯菊酯和高效氯氟氰菊酯对映体残留同时分析的GC-ECD检测方法,并应用于田间实际样品检测,结果发现本方法灵敏度高、稳定性好,能够满足茶叶和土壤中cis-联苯菊酯和高效氯氟氰菊酯手性对映体残留检测的需要。
猜你喜欢
高等学校化学学报(2024年2期)2024-03-06 06:31:12
上海农业科技(2023年4期)2023-08-07 03:05:10
食品与机械(2019年1期)2019-03-30 01:14:36
中国果业信息(2019年1期)2019-01-05 17:41:42
中成药(2018年11期)2018-11-24 02:57:32
中成药(2017年9期)2017-12-19 13:34:31
癌变·畸变·突变(2016年3期)2016-02-27 06:15:25
合成化学(2015年2期)2016-01-17 09:03:13
化工进展(2015年3期)2015-11-11 09:08:25
化学分析计量(2015年4期)2015-03-23 16:47:34
