
大肠杆菌L-苏氨酸脱氢酶的高效表达及其酶学性质分析
2023-10-21 03:10刘欣欣史红玲姚伦广唐存多
食品科学 2023年18期
刘欣欣,王 瑶,史红玲,姚伦广,王 贤,唐存多,,3,*
(1.南阳师范学院生命科学与农业工程学院,河南 南阳 473061;2.赊店老酒股份有限公司博士后创新实践基地,河南 南阳 473300;3.河南农业大学食品科学技术学院,河南 郑州 450002)
2,5-二甲基吡嗪(2,5-dimethylpyrazine, 2,5-DMP)是一种杂环含氮化合物,广泛存在于各种食品中,是食品中重要的香气化合物[1]。由于2,5-DMP具有增香作用,它作为食品工业中的食品添加剂受到了广泛关注。天然的2,5-DMP常存在于热处理后原料种子[2]、烤牛肉[3]、白醋[4]、酒[5-6]、酱油[7]、咖啡豆[8-9]、烤花生[10-11]、乌龙茶[12]以及发酵大豆[13]中,具有炒花生香气和巧克力、奶油风味,是食品工业中不可缺少的调味添加剂。同时,2,5-DMP还被应用于制药工业,是生产降糖类药物和抗脂类药物的重要底物,如5-甲基吡嗪-2-羧酸是合成抗脂分解药物中间体的合成原料[14],常用于合成抗高血压类药物阿莫西林、降血糖血脂类药物格列吡嗪以及抗结核药物5-甲基吡嗪-2-羧酸甲酯等[15-16]。
L-苏氨酸是一种必需氨基酸,可用于食品增香剂、医药、饲料添加等方面[17]。目前,安全绿色且广泛使用的生产方法是微生物发酵法[18]。近年来,L-苏氨酸的生产过热,导致出现了产能严重过剩的窘境[19]。因此,L-苏氨酸的转产增值迫在眉睫,其途径主要有以下几种:一是以L-苏氨酸为底物,经过多酶级联反应进行脱氨和还原生成L-氨基丁酸,同时还原反应所消耗的NADH通过甲酸脱氢酶再生[20]。二是以L-苏氨酸为底物,利用L-苏氨酸脱氢酶(L-threonine dehydrogenase,L-TDH)使L-苏氨酸脱氢生成L-2-氨基乙酰乙酸,经过一系列自发反应生成2,5-DMP。三是L-苏氨酸在苏氨酸醛缩酶的作用下直接生成甘氨酸。本研究拟挖掘鉴定出高活性的L-TDH,试图以L-苏氨酸为底物采用全细胞催化法生产2,5-DMP,实现L-苏氨酸的转产增值,力争解决苏氨酸产能过剩问题[12,14]。在大肠杆菌(Escherichia coli)中,2,5-DMP可以由L-苏氨酸天然合成,全细胞催化途径中的催化过程为L-苏氨酸在L-TDH的作用下转化为L-2-氨基乙酰乙酸[21],L-TDH是催化过程中的关键酶[22]。
已有文献报道,来源于E.coli的L-TDH借助pET28a载体在E.coli中实现了可溶性表达,其比活力约为132 mU/mg。曹艳丽等[19]经枯草芽孢杆菌外源表达了来源不同的L-TDH,结果表明,来源于E.coliK-12的L-TDH在枯草芽孢杆菌中更有利于L-苏氨酸合成2,5-DMP,但其L-TDH活力仅为0.024 IU,比活力为0.15 IU/mg。本研究拟以E.coli为出发菌株,从中调取L-TDH基因,再借助pACYCDuet-1质粒强化其在E.coliBL21(DE3)的表达,并对其酶学性质进行分析,旨在为提高苏氨酸转化为L-2-氨基乙酰乙酸效率提供参考。
1 材料与方法
1.1 材料与试剂
1.1.1 试剂
限制性内切酶BamH I和Hind III New England Biolabs(北京)公司;L-苏氨酸 美国Sigma-Aldrich公司;2,5-DMP、2,4-二硝基氟苯 上海阿拉丁生化科技股份有限公司;NAD+日本Chem-Station公司;异丙基-β-D-硫代半乳糖苷(isopropyl-beta-D-thiogalactoside,IPTG)Master of Bioactive Molecules(北京)公司;Prime STAR HS (Premix)、DL 2000 DNA Marker、rTaqDNA聚合酶、PrimeScript™ II 1st Strand cDNA Synthesis Kit、DNA连接试剂盒Ver.2.1 宝生物工程(大连)有限公司;甲醇、甲酸(均为色谱纯) 天津科密欧化学试剂有限公司;其他试剂均为国产分析纯。
1.1.2 菌株与质粒
本研究所涉及到的菌株和质粒如表1所示,重组质粒的构建如图1所示。

图1 重组质粒pACYCDuet-1-Ectdh的构建Fig.1 Construction of the recombinant plasmid pACYCDuet-1-Ectdh

表1 本研究用到的菌株和质粒Table 1 Strains and plasmids used in this study
1.2 仪器与设备
核酸电泳仪、Hypersil C18柱 美国Thermo Fisher公司;UNIVERSAL Hood凝胶成像系统、PowerPacTMHC/Mini-PROTEAN®蛋白电泳系统 美国Bio-Rad公司;VCX 130型超声破碎仪 美国Sonics and Materials公司;UV1000紫外-可见分光光度计 梅特勒-托利多科技(中国)有限公司;EC 2006型高效液相色谱(high performance liquid chromatography,HPLC)系统 大连依利特分析仪器有限公司。
1.3 方法
1.3.1 重组菌株的构建及序列分析
从EMBL’s European Bioinformatics Institute(https://www.ebi.ac.uk/)数据库中搜索到一个E.coli来源的L-TDHEcTDH(ACI75701.1)。根据此序列设计扩增E c t d h的特异性引物E c t d h-F:CGCGGATCCGATGAAAGCGTTATCCAAACTG( 含B a mH I 酶 切 位 点) 和E c t d h- R :CCCAAGCTTTTAATCCCAGCTCAGAATAAC(含Hind III酶切位点),委托苏州泓讯生物技术有限公司进行合成。以E.coliBL21(DE3)基因组DNA为模板,利用Ectdh-F和Ectdh-R引物进行聚合酶链式反应(polymerase chain reaction,PCR)扩增Ectdh的编码基因,再利用限制性核酸内切酶BamH I和Hind III对目的基因及载体pACYCDuet-1分别进行双酶切,然后连接转化进入E.coliBL21(DE3)感受态细胞,经氯霉素抗性筛选和测序鉴定获得E.coliBL21(DE3)/ pACYCDuet-1-Ectdh重组菌。将上述重组菌连续在无抗LB培养基上进行培养,然后接种至含氯霉素抗性的LB培养基上培养,观察生长情况,通过氯霉素抗性筛选考察重组菌携带质粒的遗传稳定性[23]。基于测序结果,通过Jalview对该酶进行多序列比对。
1.3.2 重组E.coli诱导
重组E.coli的诱导表达采用低温、低浓度诱导剂的方法。分别将E.coliBL21(DE3)/pACYCDuet-1、E.coliBL21(DE3)/pACYCDuet-1-Ectdh单菌落接种至5 mL含100 μg/mL氯霉素的LB液体培养基中,37 ℃、200 r/min振荡培养14 h,以2%接种量将过夜培养的菌液转接至100 mL新鲜LB液体培养基中,37 ℃、200 r/min培养约2 h至菌体细胞密度(OD600nm)为1.0,加入1 mol/L IPTG至终浓度为0.1 mmol/L,16 ℃、200 r/min诱导培养20 h。将100 mL菌液于8 000 r/min、4 ℃离心5~10 min,收集菌体,将菌体用20 mmol/L Tris-HCl重悬后进行超声破碎,破碎后收集上清液,上清液通过镍琼脂糖柱上进行纯化,利用十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)进行分析[24]。
1.3.3 酶活力测定
诱导表达后的重组菌进行超声破碎,离心取纯化后的上清液,参照文献[19]并进行略微改动测定重组酶活力。以L-苏氨酸为底物测定EcTDH活力,总的1 mL反应体系包含0.76 mL Tris-HCl(76 mmol/L,pH 7.8)、0.08 mLL-苏氨酸(8 mmol/L)、0.06 mL NAD+溶液(3 mmol/L)混匀,放入35 ℃金属浴中预热5 min,加入0.1 mL酶液,测定1 min内OD340nm的变化值。酶活力定义为1 min内生成1 μmol NADH所需的酶量为1 IU[25]。
1.3.4 重组酶的温度特性
参照1.3.3节方法,分别在20~60 ℃(以5 ℃为间隔)下测定重组酶活力以获得重组酶的最适反应温度。将重组酶分别在35~55 ℃保温0~120 min(以10 min为间隔),在最适反应温度下测定各自的残留酶活力,以冰浴保存各时间段的残留酶活力为100%,考察重组酶的温度稳定性。
1.3.5 重组酶的pH值特性
参照1.3.3节方法,控制在最适反应温度下,分别在pH 6.5~10.5(以0.5为间隔)下测定重组酶活力以获得重组酶的最适反应pH值。将重组酶分别在pH 6.5~10.5(以0.5为间隔)的缓冲液中放置在最适反应温度下保温1.5 h,测定各自的残留酶活力,以未经处理的酶液为100%,考察重组酶的pH值稳定性。
1.3.6 重组酶的动力学参数
根据已报道的方法稍加修改,测定重组TDH的动力学参数,确定TDH底物的特异性[26-27],反应系统中NAD+浓度为0.5 mmol/L。最佳反应条件下,在4~14 mmol/L不同L-苏氨酸浓度下测定重组酶的催化活性。每次测定重复3 次。同样地,固定L-苏氨酸浓度为10 mmol/L,将NAD+浓度设置为0.5~1.75 mmol/L,测定重组酶的催化活性,每次测定3 次,考察对NAD+的动力学参数。所有计算均由Origin 9.0执行,利用Origin 9.0软件进行非线性拟合,得出重组酶对底物的Km、Kcat和Vmax。
1.3.7 重组E.coli全细胞转化L-苏氨酸合成2,5-DMP
在250 mL三角瓶中构建50 mL的反应体系,体系包括:50 mL Tris-HCl-NaCl缓冲液、85 mmol/LL-苏氨酸、0.75 mmol/L NAD+、50 mg冻干的重组E.coli和适量玻璃珠,并于30 ℃、220 r/min振荡反应,在0~36 h内每12 h取1 mL转化液于60 ℃金属浴锅中加热10 min终止反应,于12 000 r/min离心1 min,取上清液用0.22 μm滤膜进行过滤,最后用HPLC法检测2,5-DMP含量,并绘制反应进程曲线。
1.3.8 底物和产物的检测
对底物L-苏氨酸、产物2,5-DMP进行普通C18柱的HPLC分析,HPLC色谱柱为Thermo Hypersil C18柱,检测波长为275 nm,柱温25 ℃,流动相A(体积分数0.1%甲酸)与流动相B(甲醇),按照表2进行梯度洗脱,流速为0.2 mL/min,运行时间10 min。底物L-苏氨酸HPLC检测方法通过柱前衍生测定[28-29]。

表2 HPLC梯度洗脱Table 2 HPLC gradient elution program
1.3.9 产物2,5-DMP的定量分析
将2,5-DMP标准品用超纯水分别配成1.82、2.27、3.03、4.55、9.10 mmol/L的溶液,经0.22 μm微孔滤器过滤后,取10 μL进行HPLC分析,获得各个浓度2,5-DMP的峰面积,进行线性拟合获得回归方程及相关系数。HPLC色谱柱为Thermo Hypersil C18柱,检测波长275 nm,柱温25 ℃,流动相为体积分数0.1%的甲酸和色谱级甲醇,流速0.2 mL/min,进行梯度洗脱。待检样品产物按照同样的色谱条件进行HPLC分析,根据测得的峰面积可以由回归方程计算出相应的样品浓度。
1.3.10 常用网站及软件
EMBL’s European Bioinformatics Institute(https://www.ebi.ac.uk/)数据库和Basic Local Alignment Search Tool(https://blast.ncbi.nlm.nih.gov/Blast.cgi):蛋白质、基因序列的搜索;DNAMAN软件:用于目的基因序列及限制性酶切位点分析和测序结果拼接;Origin 9.0软件:用于数据统计及分析;Jalview软件:用于酶的多序列比对;NCBI Autodock:用于分子对接。
1.4 数据统计与分析
图片均用PDF-Xchange Viewer软件进行清晰度调节,用Adobe Photoshop CS 8.0软件进行色阶、对比度调节及标注等处理。简单的数据处理及分析均借助Origin 9.0进行。
2 结果与分析
2.1 重组E. coli的构建
以E.coli的全基因组DNA为模板,Ectdh-F和Ectdh-R为引物进行PCR扩增,扩增产物进行琼脂糖凝胶电泳,如图2A所示,在约1 100 bp出现了明显的特异性条带,PCR产物片段与预期理论长度基本一致,与已报道的来源于E.coliK-12的TDH序列相似度约为77%[19]。将PCR产物用PCR产物纯化试剂盒进行纯化,用限制性核酸内切酶BamH I和Hind III进行双酶切,将酶切产物连接至经相同的限制性酶进行同样双酶切的pACYCDuet-1载体,转化至E.coliBL21(DE3)感受态细胞,再经氯霉素抗性筛选和双酶切验证,挑取重组子,用通用引物ACYCDuetUP1 Primer和DuetDOWN1 Primer进行PCR检测,结果如图2B所示,片段长度约1 100 bp,大小与其预期条带大小相符。将经PCR检测和双酶切验证的重组子送至苏州泓迅生物科技股份有限公司进行测序鉴定,获得Ectdh基因序列,并推测出其氨基酸序列。结果表明,EcTDH的序列及在载体上的插入位置与预期的结果相符,表明重组菌E.coliBL21(DE3)/pACYCDuet-1-Ectdh已成功构建。

图2 Ectdh重组子的PCR扩增(A)及pACYCDuet-1-Ectdh的通用引物验证(B)Fig.2 Agarose gel electropherograms of PCR amplified Ecctdh (A) and universal primer validation for pACYCDuet-1-Ectdh (B)
2.2 EcTDH的序列分析
将上述推测出的EcTDH氨基酸序列在ProtParam(https://web.expasy.org/protparam/)上进行理论理化性质的预测,结果表明,EcTDH的理论等电点为5.81,理论分子质量为37 173.94 Da,不稳定指数II为24.46,是一个较稳定的蛋白,其在E.coli和酵母体内的半衰期分别能达到10 h和20 h以上。利用TMHMM-2.0服务器预测蛋白跨膜结构的结果显示,EcTDH的所有氨基酸残基均为膜外残基,表明EcTDH理论上相当容易实现胞外分泌型表达。将EcTDH的氨基酸序列和已经报道来源不同的TDH氨基酸序列进行分析(激烈热球菌(Pyrococcus furiosus)、堀越热球菌(Pyrococcus horikoshii)、柯达热球菌(Thermococcus kodakaraensis)),用Jalview软件进行多序列比对,结果如图3所示,已报道的文献表明来自P.horikoshii的L-TDH氨基酸残基半胱氨酸Cys97、Cys100、Cys103、Cys111位在L-TDH中是保守的[30-31],其对应来自E.coli的L-TDH Cys93、Cys96、Cys99、Cys107。建立L-TDH的三维结构模型如图4所示,L-TDH对于底物L-苏氨酸的催化中心是在107位的半胱氨酸。

图3 EcTDH与其他3 个代表性TDH的多序列比对Fig.3 Multisequence alignment of EcTDH with three other representative TDHs
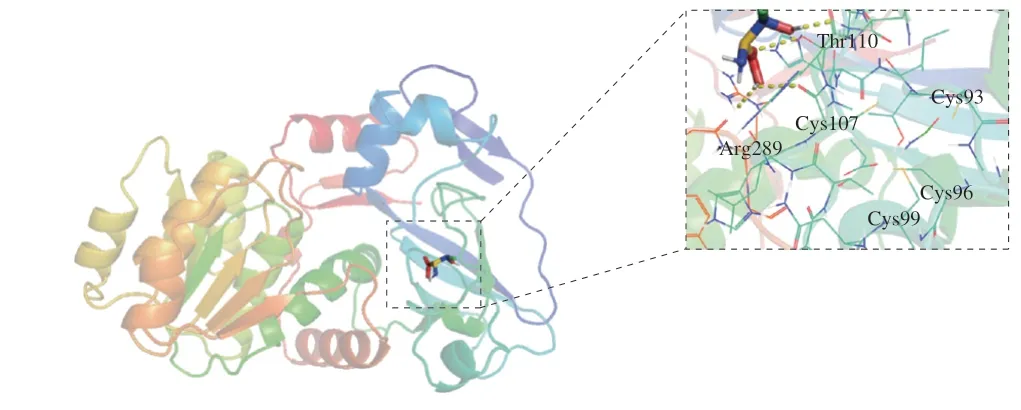
图4 EcTDH三维结构图Fig.4 Three-dimensional structure analysis of EcTDH
2.3 重组E. coli的诱导表达及鉴定
分别将E.coliBL21(DE3)/pACYCDuet-1、E.coliBL21(DE3)/pACYCDuet-1-Ectdh菌株按照1.3.2节方法进行诱导表达,将经诱导表达后的菌体进行离心,收集菌体重悬后进行超声破碎,8 000 r/min、4 ℃离心15 min后收集裂解上清液。将收集裂解的上清液进行SDS-PAGE及酶活力分析。如图5所示,E.coliBL21(DE3)/pACYCDuet-1-Ectdh裂解上清液经纯化后在约37 kDa处有明显的特异性条带,与EcTDH的理论分子质量37 173.94 Da基本一致,表明已成功实现了EcTDH在E.coli的可溶性表达。
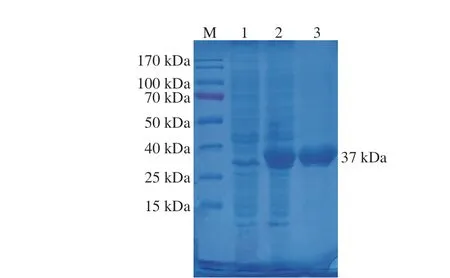
图5 重组E. coli表达产物的SDS-PAGE分析Fig.5 SDS-PAGE analysis of recombinant Escherichia coli expression products
各重组子酶活力测定结果显示,E.coliBL21(DE3)/pACYCDuet-1的酶活力为0.24 IU/mL,而E.coliBL21(DE3)/pACYCDuet-1-Ectdh的细胞裂解液中检测到EcTDH活力可达到19.13 IU/mL,与本底细胞相比,其酶活力是E.coli本底细胞表达的79 倍,纯化后的比活力可达12.77 IU/mg,表明EcTDH表现出较高的酶活力。比较已有文献报道的不同来源的L-TDH活力结果如表3所示。可以看出,来源于E.coli的L-TDH经载体pACYCDuet-1和pET28a表达后比活力产生较大差别,主要是由于酶活力定义不同,以及酶活力测定时底物和辅酶终浓度的差异导致,它们会对表观酶活造成较大的影响。文献报道[32]测定酶活力体系中底物L-苏氨酸和辅酶NAD+终浓度分别为100 mmol/L和200 mmol/L,而在本研究中终浓度分别为8 mmol/L和3 mmol/L。他们将每分钟消耗1mol NAD+所需的酶量定义为1 U,而本研究将每分钟产生1 μmol NADH所需的酶量定义为1 IU。

表3 不同来源L-TDH活力与比活力的对比Table 3 Specific activity of L-TDH from different sources
2.4 重组L-TDH的温度特性
由图6可知,在温度为20~45 ℃范围内,酶促反应速率随温度的升高而不断提高,超过45 ℃后,反应速率逐渐下降,表明EcTDH的最适反应温度为45 ℃,在40~45 ℃之间时具有较高酶活力。这与之前文献报道的最适温度一致[22]。
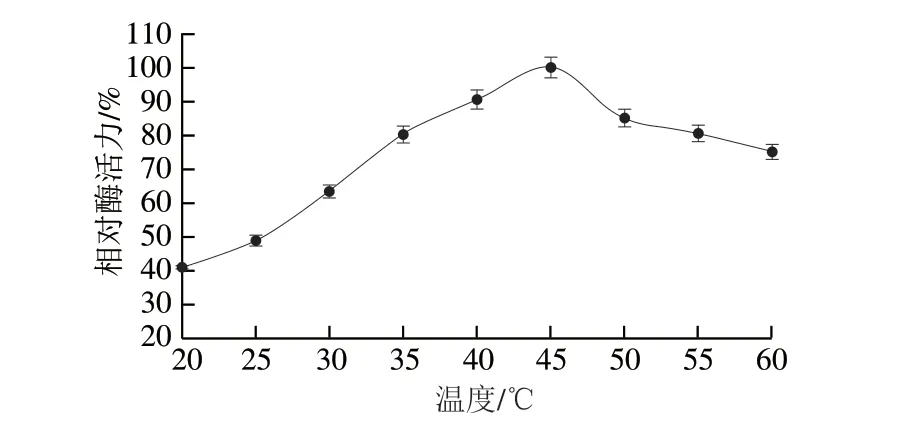
图6 重组L-TDH的最适反应温度Fig.6 Optimal reaction temperature for recombinant L-TDH
如图7所示,在35 ℃和40 ℃保温120 min后的残留酶活力仍然高于90%,这表明它在35~40 ℃范围内有较好的稳定性。与之前文献报道的在35~40 ℃条件下测定的残留酶活力相比,本研究残留酶活提高了12.5%;在45、50、55 ℃残留酶活力达到50%所对应的时间分别为78、40 min和20 min。
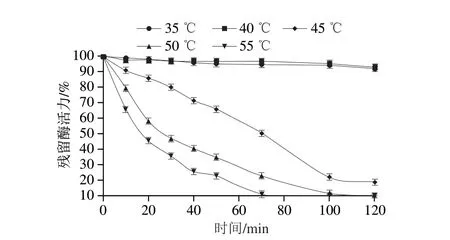
图7 重组L-TDH温度稳定性Fig.7 Temperature stability of recombinant L-TDH
2.5 重组L-TDH的pH值特性
缓冲液过酸过碱都会导致酶的空间结构的改变,从而使酶的活性降低,由图8可知,重组酶的最适反应pH值在8.5~9.5之间,相对酶活力可达80%以上,EcTDH最适反应pH值为9.0。

图8 重组L-TDH的最适pH值Fig.8 Optimal reaction pH for recombinant L-TDH
如图9所示,重组L-TDH在pH 7.5~8.5的缓冲液中保持1.5 h后残留酶活力均能保持90%以上,表明它在pH 7.5~8.5范围内有较好的稳定性,其pH值适应性相对较广,在pH 8.5~10.0范围内残留酶活力在65%以上,而已报道的L-TDH在pH值为10时,其残留酶活力仅达到60%,表明在pH值稳定性方面,该研究中L-TDH稳定性更好。
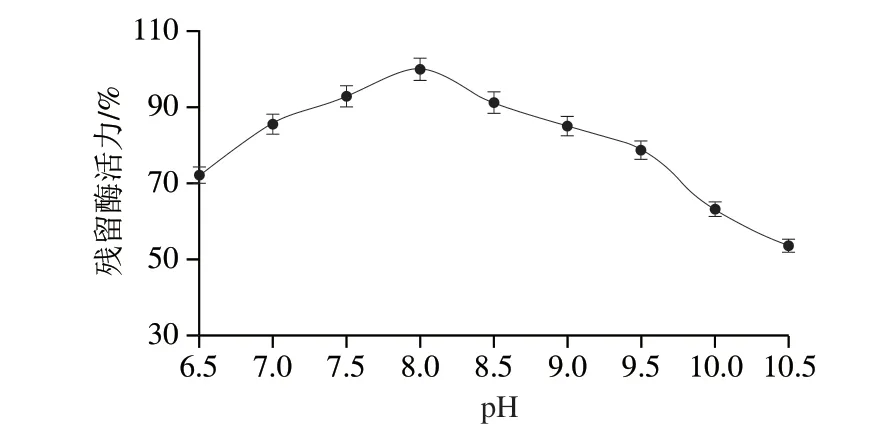
图9 重组L-TDH的pH值稳定性Fig.9 pH stability of recombinant L-TDH
2.6 重组TDH的动力学参数
表4结果显示,EcTDH对L-苏氨酸的Kcat为1.13 s-1,Km为7.76 mmol/L,Vmax为1.827 μmol/(min·mg),Kcat/Km值为0.15 L/(mmol·s)。EcTDH对辅酶NAD+的Kcat为1.52 s-1,Km为1.61 mmol/L,Vmax为2.46 μmol/(min·mg),Kcat/Km值为0.94 L/(mmol·s)。对比文献来源于Cupriavidus necator的野生型L-TDH和各位点突变型L-TDH的Km和Kcat值(表4),L-TDH因其空间结构的不同,进而影响反应过程中活性位点残基的运动使得动力学参数产生差异。

表4 不同L-TDH动力学参数的比较Table 4 Comparison of kinetic parameters of L-TDH from different sources
2.7 2,5-DMP及底物标准品的HPLC分析
按照1.3.9节方法,将分析纯的2,5-DMP在Thermo Hypersil C18柱上进行HPLC分析,在该分析条件下,2,5-DMP的保留时间约为6.55 min,将2,5-DMP不同浓度对相应的峰面积进行线性拟合,获得2,5-DMP的标准曲线,得出2,5-DMP浓度对应的回归方程为y=2 062.4x+354.29(x为2,5-二甲基吡嗪浓度(mmol/L);y为峰面积,相关系数为0.997 8),结果表明在该HPLC分析条件及浓度范围下,2,5-DMP浓度与峰面积呈良好线性相关。因此,利用此法可以对2,5-DMP进行精确定量分析。同样条件下对底物L-苏氨酸进行HPLC分析,底物的保留时间约为3.06 min。在相同的分析条件下,L-苏氨酸的保留时间与2,5-DMP保留时间差约3.5 min,结果表明在该色谱条件下能够实现L-苏氨酸与2,5-DMP的良好分离,该色谱方法能够用于催化产物的分析鉴定。
2.8 重组E. coli全细胞催化L-苏氨酸合成2,5-DMP
按照1.3.7节方法对L-苏氨酸进行全细胞催化的生物转化。E.coliBL21(DE3)/pACYCDuet-1和E.coliBL21(DE3)/pACYCDuet-1-Ectdh的催化产物经过离心取样后按照1.3.8节方法进行HPLC分析,保留时间约为6.55 min和3.06 min处有明显的特征峰,它们与相同分析条件下2,5-DMP和L-苏氨酸标品的保留时间一致,表明在该催化条件下,部分L-苏氨酸已被转化成2,5-DMP。在不同时间取样分析、计算各时间节点的产物得率,它们的催化反应进程曲线如图10所示。反应进行到36 h,反应基本达到平衡状态,2,5-DMP得率基本不再随时间的变化而变化,E.coliBL21(DE3)/pACYCDuet-1-Ectdh的产物得率约为12%,显著高于E.coliBL21(DE3)/pACYCDuet-1的转化水平。结果表明,强化L-TDH的表达可显著提高2,5-DMP合成效率,但仅依靠强化TDH的表达难以实现L-苏氨酸的高效转化。
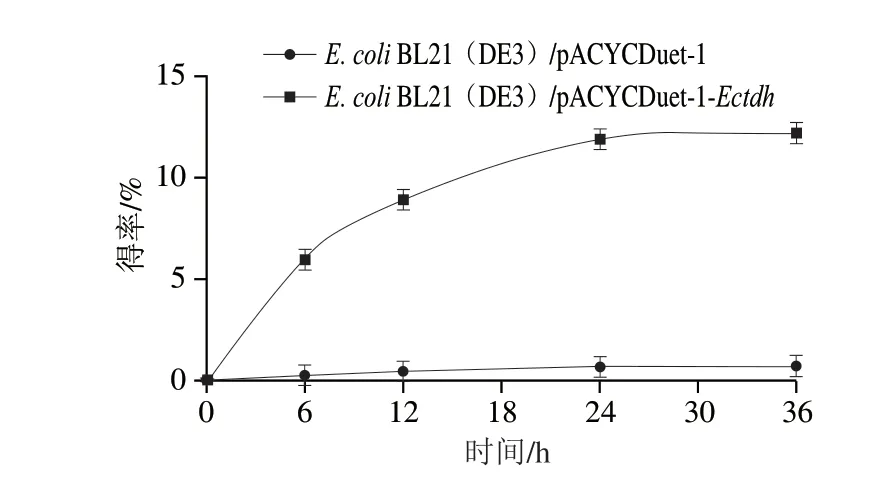
图10 L-TDH对催化反应进程的影响Fig.10 Effect of L-TDH on the catalytic reaction process
3 讨 论
L-TDH是以L-苏氨酸为底物合成高值化合物2,5-DMP的关键酶,本研究对挖掘出来源于E.coli的L-TDH进行了高效表达及酶学性质分析。本研究从E.coli中克隆出L-TDH的基因片段,再借助pACYCDuet-1表达质粒在E.coliBL21(DE3)中实现了高水平的可溶性表达,酶活力测定结果表明,E.coliBL21(DE3)/pACYCDuet-1-Ectdh发酵液中L-TDH活力可达19.13 IU/mL,纯化后重组L-TDH的比活力为12.77 IU/mg。与已报道文献相比[19],本研究表达水平和比活力均较高,比活力的差异可能归因于蛋白序列本身的差异,两者的序列相似度为77%;而表达水平的差异可能主要归因于宿主的差别,所用的E.coli表达系统比枯草芽孢杆菌表达系统具有更大表达潜力。也利用强化了TDH表达水平的重组E.coliBL21(DE3)/pACYCDuet-1-Ectdh进行了转化L-苏氨酸合成2,5-DMP的初步研究,它利用强化的TDH以及自身本底表达的NADH氧化酶、苏氨酸转运蛋白和氨基酸氧化酶等能够协同催化2,5-DMP的合成,反应24 h后产物得率约12%,显著高于没有强化TDH表达的E.coliBL21(DE3)/pACYCDuet-1,这说明TDH确实在转化L-苏氨酸合成2,5-DMP的过程中起关键作用。然而,仅强化TDH表达难以实现L-苏氨酸的高效转化,后续研究需要参考前人的研究[32],充分利用代谢工程和酶工程的手段,继续强化2,5-DMP积累正相关的基因表达,敲除2,5-DMP积累负相关的基因,以期进一步提高2,5-DMP的得率。
猜你喜欢
云南化工(2021年6期)2021-12-21
科学(2020年2期)2020-08-24
当代陕西(2020年9期)2020-08-04
饲料工业(2017年8期)2017-04-05
广东饲料(2016年1期)2016-12-01
中国工程咨询(2016年6期)2016-01-31
中国卫生(2015年7期)2015-11-08
生物技术通报(2015年1期)2015-04-10
饲料博览(2015年4期)2015-04-05
浙江人大(2014年1期)2014-03-20
