
融合生物学通路的变分自编码器在肺癌蛋白与代谢组学数据中的应用研究*
2023-10-18 14:03刘芝霖荣志炜俞轶培邱满堂侯艳
中国卫生统计 2023年4期
刘芝霖 荣志炜 俞轶培 邱满堂 侯艳,4△
【提 要】 目的 本研究提出了融合生物学通路的变分自编码器(variational auto encoder,VAE),对蛋白质与代谢组学数据进行整合分析,并应用于肺腺癌患者探索可能的病理机制。方法 为VAE的隐变量层节点赋予通路的实际意义,解码器按通路包含的生物学分子信息构建稀疏神经网络,使通路节点只与自身包含的分子连接,将隐变量作为提取的高级特征。对隐变量进行Kmeans聚类分析并使用调整兰德系数评估效果,引入基因差异表达分析方法limma探索差异表达通路,在北京大学人民医院胸外科肺腺癌患者的蛋白质与代谢组学数据中进行实例分析。结果 融合生物学通路的VAE提取的高级特征不仅将聚类准确度提高了38%,还通过差异表达分析鉴别出了实性与亚实性结节肺腺癌间的差异通路。结论 融合生物学通路的VAE可用于组学数据整合分析,其提取的高级特征具有通路表达活性的实际生物学意义。
随着各种组学测序技术的发展,产生了基因组、转录组、蛋白组、代谢组等各种组学数据,这些数据从不同组学层面描述了癌症的异质性,有利于癌症亚型分类研究及生物标志物探索,对于癌症的早期诊断、临床治疗和预后都有重要意义[1]。变分自编码器(variational auto encoder,VAE)[2]作为一种深度学习模型,具有组合低级特征为抽象的高级特征的功能,其强大的建模非线性数据关系的能力在组学数据分析中显示出极大的优越性,但缺乏解释性的问题也成为其在医学领域应用的一大阻碍[3],需要新的设计以提高模型的解释性。因此,本文提出融合生物学通路的VAE[4],将通路信息内置于神经网络的架构中,使模型提取的高级特征同时具有表征通路整体状态的实际意义。在早期肺腺癌患者的蛋白质与代谢组数据的实际应用中,揭示了亚实性结节与实性结节肺腺癌及癌旁样本间的生物学关系,为癌症机制研究提供了有生物学意义的分析结果。
原 理

ELBO=Eq(z|x)(logp(x|z))-KL(q(z|x)‖p(z))
(1)
其中q(z|x)用于近似后验分布的变分分布,p(x|z)是隐变量生成样本的条件分布,KL表示KL散度(Kullback-Leibler divergence)[6],是衡量两个分布差异的一种度量,这里用于量化分布q(z|x)与先验分布p(z)间的差异。ELBO的第一项可以通过小批量数据的蒙特卡罗抽样得到,若训练神经网络的批次大小为M,即抽样M次,则该项计算如下:
(2)
当样本生成的p(x|z)服从正态分布时,最大化该项等价于最小化重构数据的均方误差。为了能够在训练中应用标准的反向传播,需要使用重参数化技巧,假设q(z|x)服从正态分布:
q(z|x)=N(μ(x),Σ(x))
(3)
其中Σ(x)为单位对角矩阵,μ(x)和Σ(x)由编码器学习得到,然后在q(z|x)中采样,计算:

(4)
隐变量作为高级特征通常没有实际意义,这也是VAE等深度学习模型被称为“黑箱”的原因之一,缺乏生物学上的解释性限制了其应用场景。本研究为隐变量赋予了通路整体表达状态的实际意义,设定每个隐变量节点代表一个通路,使隐变量节点与生物学实体相对应,根据通路中是否包含某一生物学分子决定隐变量是否连接某一特征。这种稀疏连接的方式,能够让每个隐变量节点只汇聚指定特征的信息,强制其表征一组分子共同作用的情况,提高了模型的解释性(图1)。
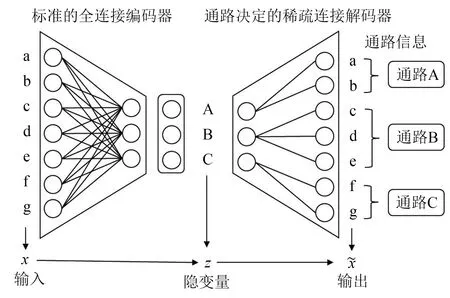
图1 变分自编码器结合生物学通路
AE也是由编码器解码器组成,但AE得到的隐变量分散在多块不连续的低维流形上,而VAE假设隐变量服从先验分布p(z),其隐变量空间平滑连续[2],更符合生物学通路表达的实际情况。解码器作为数据生成过程的条件分布p(x|z)能够解析数据的结构,而编码器用于近似q(z|x),需要足够复杂的神经网络的强大拟合能力,因此本模型只在解码器上使用了稀疏连接的结构设计。此外,由于生物学通路信息涉及多个层级的组学分子相互作用,在有合适通路信息的情况下,本模型能适用于多个组学的数据的整合分析。
对于稀疏连接神经网络的具体构建,本研究构造了一个01掩码矩阵M来实现,如果通路i中包含分子j,则Mi,j为1,否则为0。在网络的前向传播与反向传播中,网络的权重都会乘以这个掩码矩阵,使得掩码为0的权重无论是多少都强制为0,而且梯度也只在通路中的指定特征上计算,从而实现代表通路的隐变量只与通路内分子特征连接的效果。在生物学通路信息中,有的分子可能具有多种功能,从而包含在不同的通路中,为了使其在各通路中都被学习,采用神经网络的暂退法(dropout)与随机梯度下降,每次训练只使用少量挡本计算梯度并随机丢弃一些节点,在提高神经网络泛化能力[7-8]的同时也使每个包含该分子的通路都充分学习了其信息。在实际数据中训练时,由于在生物学上的认知有限,存在一些不属于任何已知通路的特征分子,因此设定了少量称为“缺失通路”的隐变量节点来连接这些特征,保证了数据能够较好的重建,同时也作为额外的通路信息以解释更多的数据变异。此外,为了提高模型的泛化能力,减少对高维组学数据的过拟合,除了使用dropout与随机梯度下降,也需要根据实际数据控制编码器解码器网络层数与节点数以调整模型复杂度。
当探索不同标签的样本间差异时,特征分子或通路表达上的变化值得关注,它们是癌症机制研究的重要线索,需要使用差异表达分析的方法。由于隐变量一般被假设为正态分布,其学习到的参数中μ作为均值代表了该通路的平均表达情况,本研究从分析差异基因表达的线性回归方法R包limma[9]得到启发,将其运用到通路表达的差异分析上,判断通路表达状态在两组间是否存在差异。此外,提取的隐变量亦可以用于其他的下游分析,如降维可视化、分析与生存时间的关联等。本研究使用的模型由python 3.9及pytorch深度学习模块搭建,隐变量的统计分析及作图使用R 4.1.3完成。
实例分析
1.实验数据集来源与整理
本研究使用来自北京大学人民医院胸外科早期肺腺癌患者的蛋白组与代谢组数据,其标签是医生通过CT影像表现确定的,分为实性结节肺腺癌、亚实性结节肺腺癌与癌旁正常组织共三类样本。组学数据由非标记定量蛋白质组学和非靶向代谢组学方法测定,所有测定均经过患者的知情同意。获得数据后,首先将蛋白与代谢物的名称转换为通路数据库中的ID,然后对数据进行标准化。排除只有一个组学信息的样本,将两个组学矩阵拼接,得到85个患者与3240分子特征的矩阵,其中实性结节肺腺癌样本37个,亚实性结节肺腺癌样本18个,癌旁正常组织样本30个;蛋白质分子特征2946个,代谢物分子特征294个。通路信息来自京都基因与基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)数据库[10],通过R包KEGGREST获取人类的通路,其名称由hsa加五位的数字编码构成,排除无关的通路后,得到77条通路信息用于设定通路节点与特征分子的连接情况。
2.实例分析结果
为了可视化隐变量对数据的分类效果,使用统一流形逼近和投影(uniform manifold approximation and projection,UMAP)方法对隐变量进行降维,该方法能够将高维数据降维并保留点之间的相互关系,本研究使用R包umap采用默认设置完成这一过程。在降至2维后,由图2可以发现正常的癌旁组织与癌症组织的差异较大,亚实性与实性的肺腺癌差异虽小,但还是明显分为了两部分,表明该方法提取的特征即使是对于早期癌症也能显示出差异。

图2 显示样本分型的隐变量UMAP图
使用Kmeans聚类方法进行无监督聚类,比较特征提取前后的变化,以评估特征提取效果。评估指标为调整兰德系数(adjusted rand index,ARI),其用于比较聚类结果与真实结果的差异,取值范围为0~1,越接近1则表明聚类效果越好。当分类为三类时,ARI在原始数据上为0.45,在隐变量上为0.62,提高了38%。当分类仅为两类,即癌与非癌时,ARI由原始数据上的0.77,提升为隐变量上的0.86,增幅为12%。可见隐变量对特征进行了优化,能够更好地对不同组织类型进行表征。
使用基于线性回归的差异表达分析方法limma对较难区分的实性与亚实性样本进行差异表达分析。对于原始数据,虽然也能分析出差异分子特征如SCEL、UBA6等,但这些分子未能在KEGG中显著富集,在生物学上也没有文献表明与癌症有明显关联,该结果解释性较差。隐变量差异分析结果的火山图(图3)显示hsa00010、hsa00630等代谢通路的表达状态上呈现显著差异,P值最小的hsa00010、hsa00630,与其他通路相比更可能与癌症代谢变化相关,而且该结果由合并通路中所有分子状态得到,具有更高的可信度。
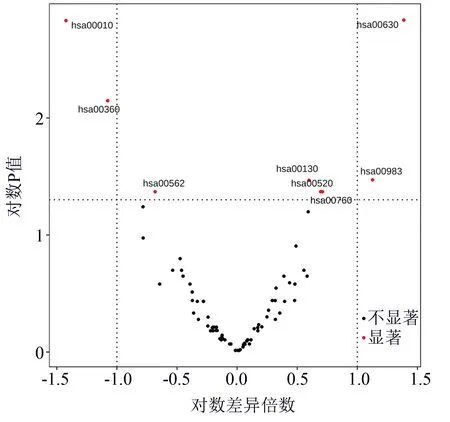
图3 隐变量差异表达分析火山图
hsa00010是与糖酵解与糖异生相关的通路,其调控关系如图4所示,实性结节肺腺癌与亚实性结节肺腺癌相比,红色表示上调,绿色表示下调,白色表示因目前技术局限性未测定的分子。由于通路中大部分分子的表达差异在统计学上并不显著,但将它们当作整体分析时若呈现一致的变化可能会使得通路整体活性表现出统计学差异,为了更好地呈现这种趋势,上调和下调仅根据表达均值计算。从图中可以看出大部分分子呈现上调的趋势,而且在通路调控关系中距离较近,能够相互影响作用。该结果表明,在分析单个分子的差异时难以发现的一系列分子微小同步变化,累积起来会有不小的效应,导致通路整体表达水平的变化,直接将通路作为整体分析能够较好地发现这种趋势的差异。

图4 实性结节肺腺癌相较于亚实性结节肺腺癌在糖酵解/糖异生通路的变化
hsa00630是乙醛酸和二羧酸代谢通路(图5),通路里的大量分子也显示出同步上调,带来通路整体水平的变化,然而其在火山图里呈现出与hsa00010不同方向的变化,这是因为作为通路状态的高级特征值的上升不一定代表通路活性的上调,高级特征的提取经过了神经网络的非流形转换,值的高低不等于通路活性的高低,但值的差异却可以反映通路活性的差异。
讨 论
本研究提出了一种新的VAE架构,采用已知的生物学通路信息构建稀疏解码器,以此分析不同样本在通路水平上的活动情况。融合生物学通路信息的VAE能够将高维的组学数据按照通路信息有序整合为各个通路整体表达水平,提高了表征样本的能力,同时便于在通路的层级上进行差异表达分析,探索疾病机制。对于这一思路衍生的其他模型,如使用AE代替VAE或在编码器中使用稀疏连接,实际测试表明它们的效果不如当前的模型,与理论分析的结果一致,最终本研究采用了稀疏连接编码器的VAE。牺牲小部分数据重建能力,以获得更有意义的高级特征,这对于医学方面的应用更加重要,而且未来更全面的通路信息也许能够实现比全连接更好的数据重建。此外,本研究使用了通路信息作为生物学先验知识构建神经网络,其可以推广到调控网络、生物学模块等多种生物学概念上,根据包含的生物学分子构建多种多样的连接方式,具有广泛的应用前景。
在癌症的发生发展中,为了满足肿瘤细胞的生物能量、生物合成和氧化还原需求,会对代谢通路的调控机制进行重编程,导致异常的通路活性[11],所以代谢通路的改变在癌症机制分析中十分重要,能够发现重要的生物标志物,这一点在肺腺癌中也得到了证实[12]。以前的研究也发现亚实性结节的肺腺癌相对于实性结节的具有更好的生存率[13],本研究也得到了相似的结论:基于现有数据的通路活性的差异分析表明,实性结节的样本中能量相关代谢通路比亚实性结节样本有更高的活性,可能是导致其进展更快的原因。目前在蛋白质与代谢组学上对实性与亚实性肺腺癌的研究还较少,本文能够为相关的机制研究提供可能的线索。
本研究使用的数据来自早期癌症的患者,所以数据本身的区分度还不够大,特别是在实性与亚实性间,但本方法还是能够发现一些与致癌机制相关的线索,提升了样本间的区分度,证明了方法的有效性。传统的差异分析方法先分析出差异特征,再根据这些特征做通路富集,然而这种做法存在一定缺陷。本研究中,分析原始数据得到的差异分子不是每一个都得到富集;同时,从隐变量中得到的差异通路包含的分子本身并没有显示出统计学上的差异,这些问题都降低了结果的解释性。本研究发现。一系列同步的“不显著”变化能够导致通路总体表达水平的显著变化[14],因此以通路为整体分析在生物学上更合理,错误率也更低[15]。在癌症等疾病早期患者中,相关的分子表达还未能显现出差异,但通路内的分子若出现同步微小变化,就能在通路水平上被捕获到,这对于早期诊断具有重要意义。
组学数据分析方法不仅需要适应数据的复杂性与异质性[16],还要有良好的生物学解释性。深度学习方法拟合能力强但解释性较差,基于通路的分析错误率低且生物学解释性好,融合生物学通路的VAE结合了两者的优势,对多组学数据的整合分析和精准医学的发展都能起到重要作用[17]。
猜你喜欢
中国临床医学影像杂志(2022年5期)2022-07-26
国际口腔医学杂志(2019年3期)2019-05-31
中成药(2018年7期)2018-08-04
天然产物研究与开发(2018年2期)2018-04-04
中外医疗(2016年15期)2016-12-01
西南军医(2016年3期)2016-01-23
肿瘤预防与治疗(2015年5期)2015-09-26
医学研究杂志(2015年11期)2015-06-10
中国当代医药(2015年17期)2015-03-01
西南军医(2014年5期)2014-04-25
