
靶向RelA(p65)基因shRNA慢病毒载体构建及功能鉴定
2023-10-18 07:14:08霍云飞高明慧豆双双寇卜心柴梦音刘晓霓
中国临床新医学 2023年9期
霍云飞, 高明慧, 豆双双, 寇卜心, 柴梦音, 刘晓霓, 石 英
RelA(p65)是核因子κB(nuclear factor kappa-B,NF-κB)家族的一员。NF-κB通路包括典型的NF-κB通路和非典型的NF-κB通路,其中RelA作为重要的转录因子参与典型的NF-κB通路[1-2]。在稳定状态下,RelA被NF-κB抑制蛋白(inhibitor of NF-κB,I-κB)抑制,隔离在细胞质中。当典型的NF-κB通路被激活后,I-κB被磷酸化,失去了抑制RelA的能力,RelA进入细胞核中参与下游基因的转录,以响应炎症、免疫反应、细胞增殖、分化等多种外部刺激[1,3-5]。此外,也有研究表明RelA参与了恶性肿瘤的发生、发展,特别是在肝癌中发挥重要作用。Xu等[6]发现当肝脏发生慢性炎症反应后,肝细胞RelA表达上调,并激活RelA的Ser536磷酸化位点,继而激活下游丝氨酸-苏氨酸激酶(serine-threonine protein kinase,Akt)/哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)通路,促进肝癌的发生、发展。本研究团队的前期研究也发现,RelA的磷酸化促进了小鼠肝癌的发生[7]。因此,抑制RelA的表达可能可以抑制肝癌的进展。RNA干扰(RNA interference,RNAi)是指在进化过程中高度保守的、由双链RNA(double-stranded RNA,dsRNA)诱发的、同源mRNA高效特异性降解的现象[8]。短发夹RNA(short hairpin RNA,shRNA)表达载体的优点在于可以进行较长期研究——带有抗生素标记的载体可以在细胞中持续抑制靶基因的表达,持续数周甚至更久[9]。慢病毒载体也可用于小分子干扰RNA(small interfering RNA,siRNA)表达,其优势在于可以直接高效率感染细胞以进行基因沉默的研究,避免由于质粒转染效率低而带来的种种不便,而且转染效果更加稳定[9]。本研究旨在构建靶向RelA基因的慢病毒载体,抑制肝癌细胞中RelA的表达,为后续RelA在肝癌发生、发展中的机制研究奠定基础。
1 材料与方法
1.1主要试剂 质粒小量快速提取试剂盒购自北京艾德莱生物科技有限公司;限制性核酸内切酶购自美国Thermo公司;慢病毒包装试剂盒购自北京和元生物科技有限公司;细胞基因组DNA提取试剂盒购自天根生化科技有限公司;RNA提取试剂盒Fast Pure Cell/Tissue Total RNA Isolation Kit V2购自南京诺唯赞生物科技股份有限公司;逆转录试剂盒PrimeScriptTMII 1ST Strand cDNA Synthesis Kit、荧光定量聚合酶链式反应(polymerase chain reaction,PCR)试剂盒TB Green Premix Ex TaqTM购自日本Takara公司;DMEM培养基、胎牛血清购自美国Gibco公司;遗传霉素G418购自碧云天生物科技有限公司;GAPDH抗体、RelA抗体购自美国Cell Signaling Technology公司;CCK-8试剂购自美国AbMole公司。
1.2慢病毒载体及细胞 慢病毒载体pCLenti-U6-shRNA-CMV-mCherry-F2A-Neo-WPRE购自上海和元生物科技有限公司。HepG2细胞及293T细胞由本实验室保存,均用含10%胎牛血清的DMEM培养基进行培养,培养箱条件设置为37 ℃、5% CO2。
1.3主要实验仪器 MCO-18AIC恒温二氧化碳细胞培养箱购自日本SANYO公司;Ti-E全电动倒置荧光显微镜、Ts2倒置显微镜均购自日本Nikon公司;EPS-3000电泳仪购自上海TANON公司;T100 PCR仪购自美国BioRad公司;ViiA7实时荧光定量PCR仪购自美国ABI公司;MULTISKAN GO全波长酶标仪购自美国Thermo公司。
1.4实验方法
1.4.1 目的基因干扰靶点的设计 根据美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)中RelA基因的核酸序列,设计siRNA靶向序列和阴性对照序列。见表1。
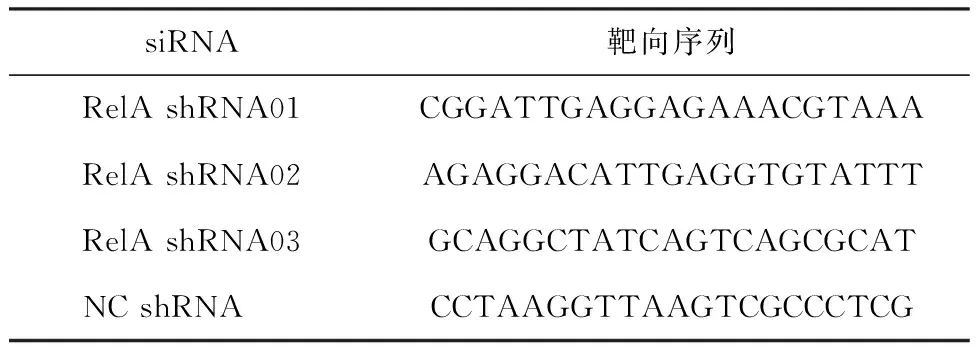
表1 siRNA靶向序列
1.4.2 shRNA的合成及退火 shRNA由上海和元生物科技有限公司进行合成。在合成的shRNA中,loop茎环结构为CTCGAG,3′端加入TTTTTT终止信号,5′端和3′端分别加入Age I和EcoR I酶切位点。见表2。将合成的单链shRNA用oligo annealing buffer溶解成20 μM,互补单链各取30 μl进行混合,置于95 ℃水浴锅中加热5 min,然后室温冷却,形成双链oligo片段。取1 μl双链oligo片段用于后续的连接反应,其余-20 ℃保存。
1.4.3 线性化表达载体的制备 取2 μg载体质粒pCLenti-U6-shRNA-CMV-mCherry-F2A-Neo-WPRE,在Age I和EcoR I酶的作用下于37 ℃水浴锅中孵育2 h进行酶切,使其线性化。通过琼脂糖凝胶电泳验证酶切效果并回收大小约为8 688 bp的正确条带。
1.4.4 载体质粒与退火的shRNA连接 将上述线性化表达载体与退火的双链oligo序列的连接反应体系(见表3)于16 ℃过夜连接,完成慢病毒基因沉默质粒构建。见图1。
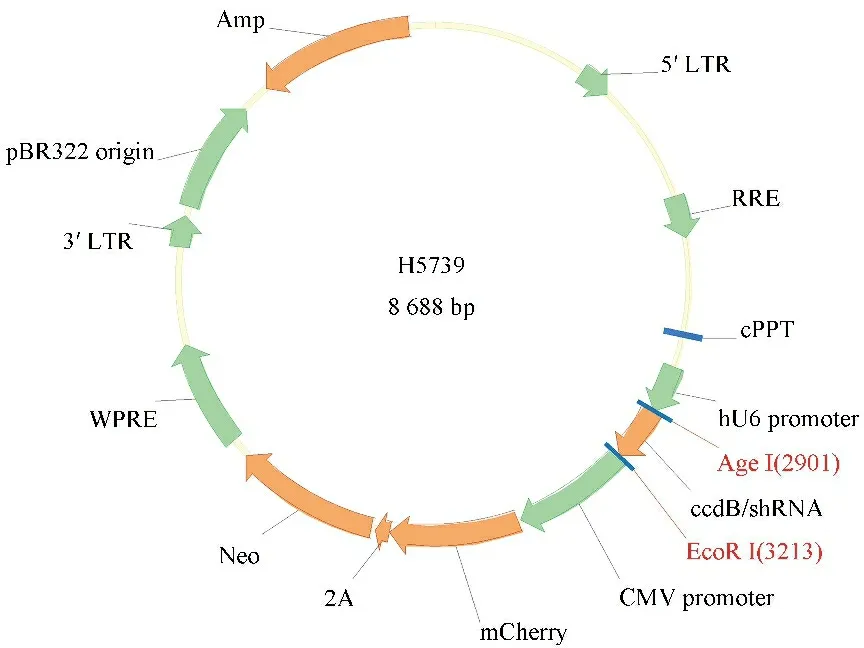
图1 RelA基因沉默慢病毒质粒示意图

表3 连接反应体系
1.4.5 转化、菌落PCR鉴定及测序 用构建完成的RelA基因沉默慢病毒质粒转化DH5α感受态细胞,涂布于含有氨苄西林抗性的LB固体培养基平板,37 ℃培养过夜。挑取平板上长出的转化子重悬于10 μl LB培养液中,取1 μl作为模板进行菌落PCR鉴定,并将得到的阳性克隆进行测序验证(测序引物序列:F:TACGATACAAGGCTGTTAGAGAG;R:CTATTAATAACTAATGCATGGC)。经测序验证正确的阳性克隆,用质粒小量快速提取试剂盒提取质粒,将提取出的质粒用于慢病毒的包装。
1.4.6 慢病毒包装 接种5×106cells 293T细胞于10 cm培养皿中,过夜培养后使用慢病毒包装试剂盒进行慢病毒包装系统转染。转染48 h后,收集上清,与浓缩试剂按照5∶1的比例在4 ℃温度下进行混合,过夜,离心收集沉淀并使用PBS重悬,即得到病毒浓缩液,-80 ℃保存。
1.4.7 慢病毒滴度测定 接种1×105cells 293T细胞于12孔板中,过夜培养后进行病毒滴度测定。分别取0.1 μl、1 μl、10 μl病毒加入到12孔板中,同时添加终浓度为8 μg/ml的促感染试剂Polybrene。培养72 h后拍照记录病毒感染后的情况并利用细胞基因组DNA提取试剂盒提取基因组DNA,用于实时定量PCR(quantitative real-time PCR,qRT-PCR)检测。根据公式TU ml-1=(C×N×D×1 000)/V计算出每毫升慢病毒液中含有的具有生物活性的病毒颗粒数,即为慢病毒滴度,C为平均每基因组整合的病毒拷贝数;N为感染时细胞的数目;D为病毒载体的稀释倍数,V为加入的稀释病毒的体积数(μl)。
1.4.8 慢病毒转染HepG2细胞及干扰效果鉴定 接种1×105cells HepG2细胞于6孔板中,过夜培养后再加入慢病毒和终浓度为8 μg/ml的Polybrene。每孔所加病毒量(μl)=MOI×感染时的细胞数/病毒滴度×1 000,其中HepG2的MOI值为10~30。转染24 h后更换不含慢病毒的新鲜培养基继续培养48 h,荧光显微镜下观察感染情况并拍照记录。更换含有遗传霉素G418的新鲜培养基筛选转染成功的细胞,当未转染慢病毒的细胞在含有遗传霉素的培养基中全部死亡后即可认为所有细胞均感染成功。收集转染成功的细胞,通过Western blot实验观察RelA基因在蛋白质水平上的表达情况,筛选干扰效果最好的细胞,通过CCK-8法检测细胞增殖活力,增殖活力=(A1-Ab)/(A0-Ab),A1为细胞培养24 h、48 h和72 h后测得的OD450值,A0为细胞培养0 h时测得的初始OD450值,Ab为空白对照孔的OD450值。
2 结果
2.1RelA基因沉默慢病毒质粒载体构建及鉴定结果 在Age I和EcoR I酶的作用下,载体质粒pCLenti-U6-shRNA-CMV-mCherry-F2A-Neo-WPRE被切割成线性化载体,琼脂糖凝胶电泳显示环状载体和切割后的线性化载体在8 000~10 000 bp之间出现条带,与载体实际大小8 688 bp一致,提示线性化载体切割成功。见图2ⓐ。通过aKaRa MiniBEST Agarose Gel DNA Extraction Kit Ver.3.0试剂盒进行胶回收,得到的线性化载体在T4 DNA ligase的作用下与退火的双链oligo序列进行连接,然后进行菌落PCR。琼脂糖凝胶电泳显示,Y4056、Y21318、Y21319、Y21320四种慢病毒载体条带大小均略低于500 bp,与实际的474 bp一致,提示成功筛选出阳性克隆。见图2ⓑ。DNA测序后通过Vector NTI软件比对,显示各重组质粒载体序列与设计序列一致。见图3。综合上述结果提示pCLenti-U6-shRelA-CMV-mCherry-F2A-Neo-WPREo质粒(Y4056、Y21318、Y21319、Y21320)构建成功。
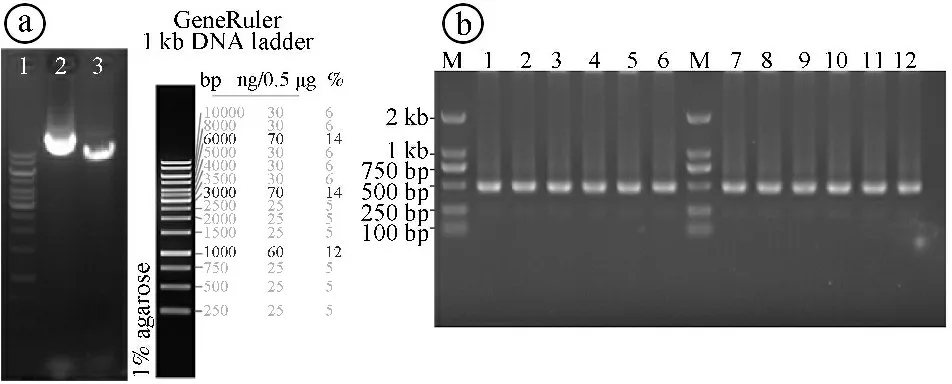
ⓐ载体酶切结果,1:DNA ladder marker;2:H5739载体;3:H5739载体酶切后片段。ⓑ菌落PCR鉴定结果,M:DL2000 DNA marker;1~3:Y21318挑取的3个转化子;4~6:Y21319挑取的3个转化子;7~9:Y21320挑取的3个转化子;10~12:Y4056挑取的3个转化子图2 RelA基因沉默慢病毒质粒载体凝胶电泳及菌落PCR鉴定结果图
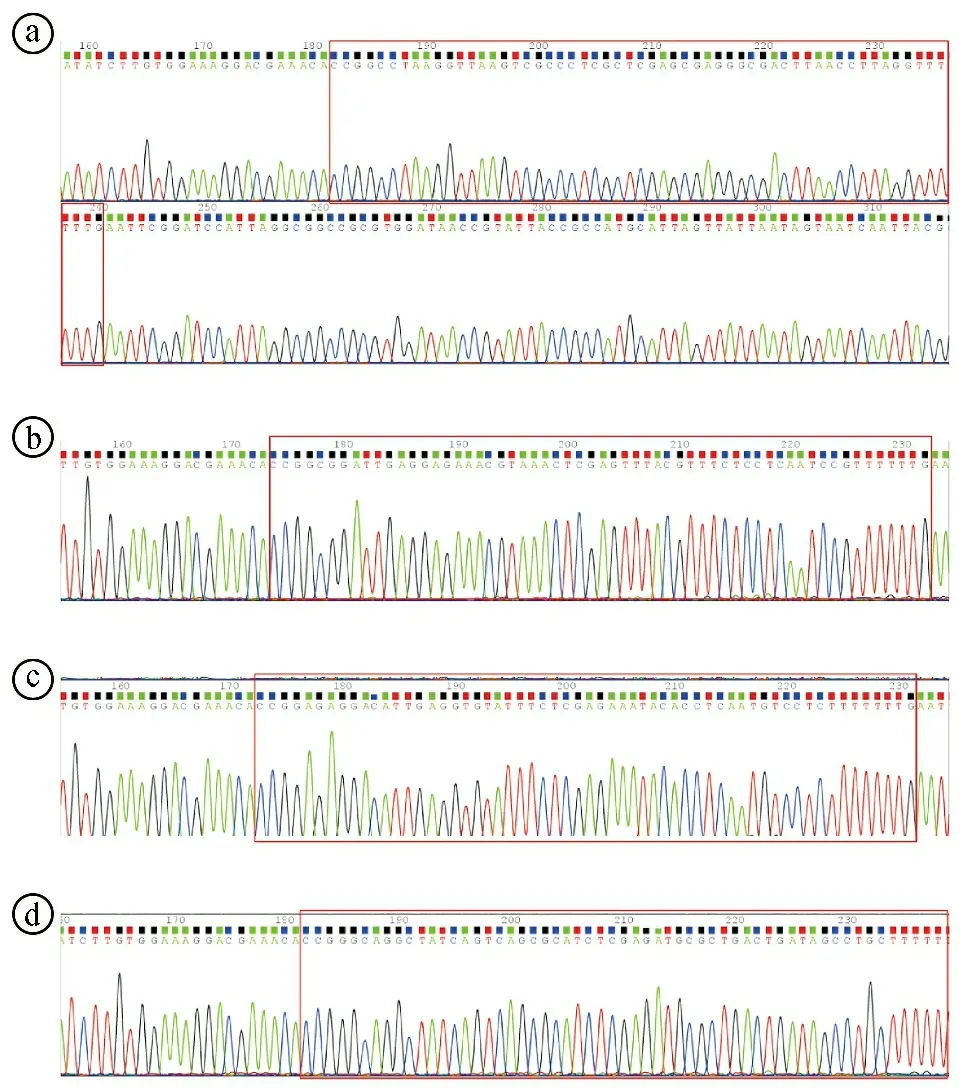
ⓐY4056;ⓑY21318;ⓒY21319;ⓓY21320图3 RelA基因沉默慢病毒质粒测序比对结果图
2.2慢病毒Lenti-shRelA包装及滴度检测结果 采用慢病毒包装试剂盒和293T细胞将基因沉默慢病毒质粒包装后并进行滴度测定。转染72 h后,荧光显微镜观察到四种慢病毒的荧光强度随病毒含量升高均逐渐升高。见图4。提取293T细胞的基因组DNA,通过qRT-PCR测定各慢病毒滴度,舍去误差较大的结果,最终得出四种慢病毒滴度分别为3.13×108TU/ml(Y4056)、2.97×108TU/ml(Y21318)、2.51×108TU/ml(Y21319)、3.40×108TU/ml(Y21320)。见表4~7。

ⓐY4056;ⓑY21318;ⓒY21319;ⓓY21320图4 慢病毒转染293T细胞荧光图(×100)
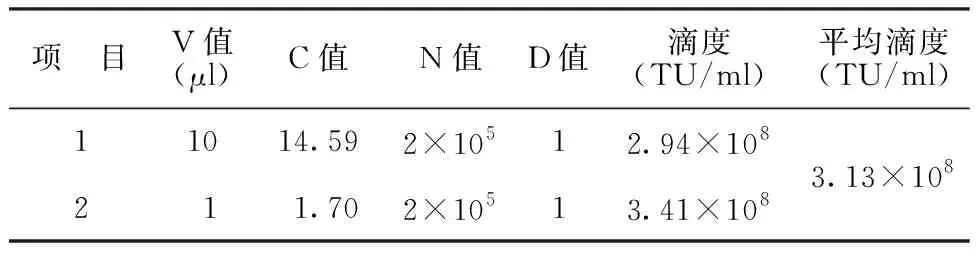
表4 Y4056滴度测定结果

表5 Y21318滴度测定结果

表7 Y21320滴度测定结果
2.3Lenti-shRelA对HepG2细胞的干扰效果 使用上述四种慢病毒转染HepG2细胞,当未转染慢病毒的细胞在含有遗传霉素的培养基中全部死亡后,荧光显微镜下观察到所有细胞均表达mCherry红色荧光,提示转染成功。见图5ⓐ。Western blot实验结果显示,发现与对照Y4056相比,Y21318、Y21319和Y21320三种慢病毒感染的HepG2细胞的RelA在蛋白质水平上的表达均显著降低(P<0.05),其中Y21320干扰效果最好。见图5ⓑⓒ。选择干扰效果最好的Y21320慢病毒进行后续实验。
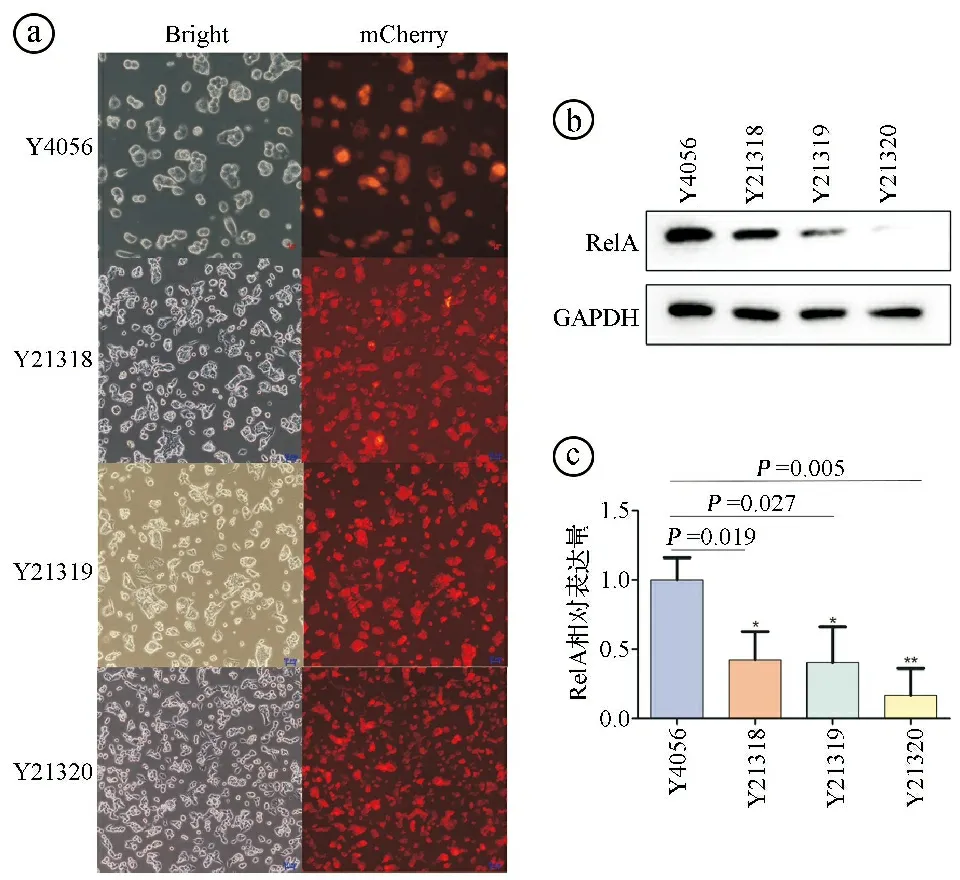
ⓐ荧光显微镜观察HepG2细胞经慢病毒感染后的mCherry红色荧光表达情况(×100);ⓑⓒWestern blot实验验证HepG2细胞经慢病毒感染后RelA蛋白的表达,与Y4056比较,*P<0.05图5 Lenti-shRelA对HepG2细胞的干扰效果图
2.4RelA敲降抑制HepG2细胞增殖 通过CCK-8实验检测RelA敲降后对细胞增殖活力的影响,结果显示,RelA敲降后细胞增殖活力降低。见图6。提示RelA在调控细胞增殖上发挥着重要作用。
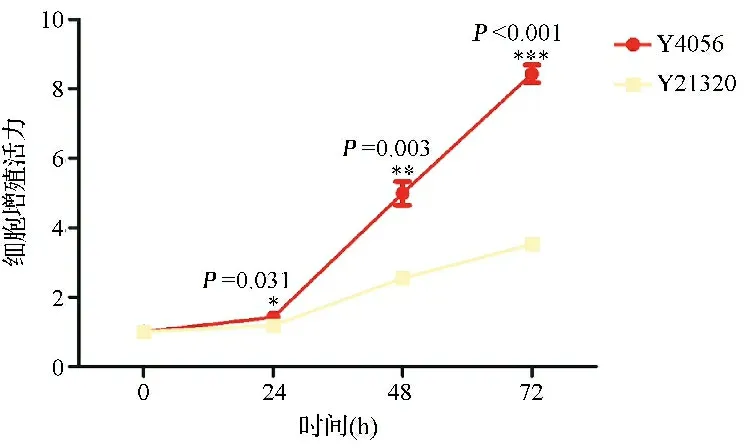
图6 RelA敲降对HepG2细胞增殖活力影响的结果图(*P<0.05)
3 讨论
3.1RNAi是指在进化过程中高度保守的、由dsRNA诱发的基因沉默现象[8]。由于shRNA表达载体可以稳定抑制靶基因,已经成为肿瘤领域常用的科学研究手段[10]。慢病毒载体是一类由人类免疫缺陷病毒1(human immunodeficiency virus type 1,HIV-1)改造的病毒载体,优势在于可以直接高效率感染细胞进行基因沉默的研究,避免由于质粒转染效率低而带来的不便,而且转染效果更加稳定[9,11]。本研究采用慢病毒载体介导的RNAi技术稳定沉默HepG2细胞的RelA基因。
3.2RelA作为NF-κB家族的一个重要的转录因子,可通过影响下游靶基因的表达调控细胞的增殖、炎症反应、免疫、分化等,对肿瘤的发生、发展具有重要作用。研究发现,在胰腺癌中,沉默含FYVE、Rho GEF和PH结构域蛋白3(FYVE,RhoGEF and PH domain-containing 3,FGD3)可上调RelA表达而促进胰腺癌细胞增殖和转移[12]。在肺癌中,NF-κB亚单位p50/p65激活可以促进肺腺癌细胞H1650对吉非替尼的耐药[13]。在乳腺癌中,RelA是F-盒和含蛋白2的WD重复结构域(F-box and WD repeat domain-containing protein 2,FBXW2)的底物,两者表达水平呈负相关。当敲降FBXW2表达后,RelA表达升高,从而促进了肿瘤的生长。在FBXW2敲降的基础上敲降p65,肿瘤的生长可被抑制[14]。
3.3RelA在肝癌的发生、发展中也发挥着重要的作用。据统计,在所有癌症的发病率和死亡率中,肝癌分别位于第6位和第3位[15],而目前对于肝癌的治疗方法,包括手术、介入、靶向药物和免疫检测点药物治疗等,仍存在疾病复发率高、患者生存率低等问题[16]。因此,寻找肝癌新的治疗靶点尤为重要。研究发现,通过抑制RelA的核易位,减少氧化应激和炎症,可改善肝纤维化[17];抑制NF-κB p65的磷酸化可以改善肝损伤,抑制肝癌细胞的增殖和侵袭[18-19];RelA表达水平升高促进了小鼠肝癌的发生[20]。上述研究结果提示抑制RelA的表达或磷酸化,可能有助于抑制肿瘤的进展。
3.4在肿瘤中某些基因的异常表达常常引起RelA表达或磷酸化水平的异常,进而影响肿瘤的增殖、迁移等[21-23]。本研究团队的前期研究发现,二乙基亚硝胺(diethylnitrosamine,DEN)可以通过激活NF-κB通路,使RelA磷酸化水平上升,诱导促炎因子的表达,从而促进肝癌的发生,而NF-κB通路抑制剂QNZ可以逆转这种作用[7]。提示RelA在肝癌的发生、发展过程中可能发挥着重要作用,但其具体生物学机制仍未被完全阐明。鉴此,本研究设计合成了RelA干扰序列并连接到H5739载体上,获得了滴度分别为3.13×108TU/ml(Y4056)、2.97×108TU/ml(Y21318)、2.51×108TU/ml(Y21319)、3.40×108TU/ml(Y21320)的慢病毒载体。实验结果显示,本研究所构建的RelA shRNA慢病毒载体在体外可高效转染HepG2细胞,经过遗传霉素G418的筛选,死亡细胞较少,且95%以上细胞呈现红色荧光。Western blot实验结果显示,shRelA慢病毒载体干扰效率较高,并筛选出了干扰效果最好的Y21320进行后续研究。CCK-8实验结果显示,敲降RelA表达可显著抑制HepG2细胞的增殖,提示RelA在调控细胞增殖上可能发挥着重要作用,但其机制仍需进一步探究。
综上所述,本研究成功构建了RelA基因shRNA慢病毒载体,获得了稳定沉默RelA基因的HepG2细胞,为进一步研究RelA基因在肝癌发展中的机制奠定了基础。
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29 00:51:20
华侨大学学报(自然科学版)(2021年4期)2021-07-30 02:18:58
中国保健营养(2021年16期)2021-04-03 18:15:56
食品科学(2018年10期)2018-05-23 01:27:28
法医学杂志(2015年4期)2016-01-06 12:36:36
法医学杂志(2015年4期)2016-01-06 12:36:36
西南医科大学学报(2015年1期)2015-08-22 13:01:46
中国当代医药(2015年9期)2015-03-01 02:01:59
现代检验医学杂志(2015年2期)2015-02-06 02:01:10
西南军医(2015年6期)2015-01-23 01:25:50
