
改造乙酰羟酸合成酶提高L-亮氨酸产量
2023-10-17 07:56:48徐建中
食品与生物技术学报 2023年9期
刘 宁, 徐建中*
(1. 江南大学生物工程学院, 江苏 无锡 214122;2. 江南大学工业生物技术教育部重点实验室, 江苏 无锡 214122)
L-亮氨酸是主要的支链氨基酸之一,主要应用于食品、医药、动物饲料和化妆品行业。 目前工业生产L-亮氨酸的主要方法是微生物发酵法、直接提取法和化学合成法[1]。 微生物发酵法由于成本低、环境友好等优点而被广泛运用,工业常用的菌株为谷氨酸棒杆菌和大肠杆菌[2]。 在谷氨酸棒杆菌中,L-亮氨酸生物合成与L-异亮氨酸和L-缬氨酸合成密切相关, 因为这3 个支链氨基酸都以丙酮酸为前体,并且代谢步骤中经过4 个相同的酶催化反应[3-4],依次由乙酰羟酸合成酶(AHAS,编码基因ilvBN)、乙酰羟酸异构还原酶(AHAIR,编码基因ilvC)、二羟酸脱氢酶(DHAD,编码基因ilvD)以及支链氨基酸转氨酶(TAs, 编码基因ilvE) 催化(见图1)[5-6]。 其中AHAS 是由两个ilvB 基因编码的大亚基(催化亚基)和两个ilvN 基因编码的小亚基(调节亚基)组成的四聚体。 它是合成途径中的第一个限速酶,其调节亚基受L-亮氨酸、L-缬氨酸和L-异亮氨酸的协同反馈阻遏, 作用原理是其能与AHAS 的调节位点结合,从而抑制AHAS 活性[7]。 在大肠杆菌(Escherichia coli) 和鼠伤寒杆菌(Salmonella typhimurium) 中,AHAS 存在3 种同工酶, 分别是AHASI、AHASII 和AHASIII, 由操纵子 ilvBN、ilvGMEDA 和ilvIH 所编码,但并不是全部的细菌都有多种AHAS 同工酶,在谷氨酸棒杆菌中就只存在一种AHAS。 在谷氨酸棒杆菌中,AHAS 主要是受L-缬氨酸的反馈抑制,并且其可以同时催化2 个反应, 第一个反应是催化丙酮酸生成2-乙酰乳酸,进入L-亮氨酸和L-缬氨酸的合成途径; 第二个反应则是催化2-酮基丁酸和丙酮酸生成2-乙酰-2-羟基丁酸,进而合成L-异亮氨酸[8-9]。因此,AHAS 在支链氨基酸的合成过程中占有重要的地位。
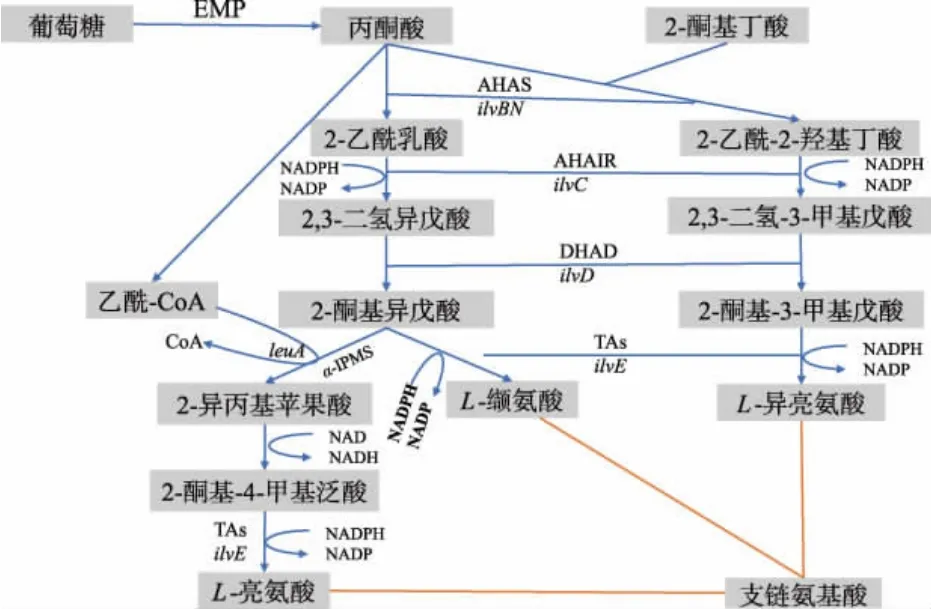
图1 L-亮氨酸合成代谢途径Fig. 1 Metabolic pathway of synthetic L-leucine
目前对AHAS 的蛋白质改造主要集中于降低其对支链氨基酸的敏感性,从而来降低支链氨基酸的反馈抑制[10]。 Guo 等发现A42V、A89V、K136E、A42V、A89V 这些突变位点对于AHAS 的蛋白质改造具有一定意义[11];Zhang 等首先过表达基因ilvBN, 然后将47 位异亮氨酸突变成酪氨酸来减少反馈抑制, 最终重组菌株的L-亮氨酸产量达到了32.1 g/L[12];Hasegawa 等发现如果将谷氨酸棒杆菌中ilvN 基因的156 位Gly 定点突变成Glu, 并且敲除谷氨酸棒杆菌中的ldhA 基因, 同时过表达ilvBNCDE 可以有效提高AHAS 抗反馈抑制的能力,结合工艺优化后L-缬氨酸产量可达227 g/L[13];Lu 等通过定点诱变技术,定点诱变AHAS 调节亚基上的位点(N11S、T34I、A36V、T104S、N11F、G14E 和N29H),使突变体对L-异亮氨酸敏感性显著下降[14]。同时,在代谢工程方面对AHAS 的改造也有一定的进展。 Hou 等在黄色短杆菌中串联表达经定点突变的ilvEBNrC 基因,L-缬氨酸产量达到了38 g/L[15];Wang 等将基因ilvBNC 的原始启动子替换成Ptuf启动子后,重组菌株最终摇瓶发酵可获得28.47 g/L 的L-亮氨酸[7];崔毅引入L-缬氨酸生产菌中的AHAS突变体, 最终分批补料发酵生产39.8 g/L 的L-亮氨酸[9];陈宁等改变中心碳代谢途径,最终使得L-亮氨酸的产量(53.0 g/L)提高10.0%[16]。
在谷氨酸棒杆菌中,AHAS 对2-酮基丁酸的亲和力明显高于丙酮酸,所以当丙酮酸和2-酮基丁酸同时存在时,AHAS 会优先利用2-酮基丁酸合成L-异亮氨酸[17]。 由此可以推测出当存在较高含量的2-酮基丁酸时, 可能会造成L-缬氨酸和L-亮氨酸的合成受限制而造成生长缺陷[18],从而不利于L-亮氨酸的积累。 作者通过蛋白质改造工程对AHAS 进行理性改造,针对其底物偏好性做出优化,利用同源建模和分子模拟找出合适突变位点,筛选出最优突变体来提高AHAS 对丙酮酸的催化能力,从而提高L-亮氨酸产量。
1 材料与方法
1.1 主要试剂
异丙基β-D-硫代半乳糖苷 (IPTG)、2×Phanta Max Master Mix、2×Tag Max Master Mix (Dye Plus)、卡那霉素: 南京诺唯赞生物科技有限公司产品;限制性内切酶、DNA 连接酶: 赛默飞世尔科技有限公司产品;酵母提取物、胰蛋白胨:英国Oxoid 公司产品;质粒提取试剂盒、基因组提取试剂盒、胶回收试剂盒、定点突变试剂盒:上海生工生物工程股份有限公司产品。
1.2 菌株与质粒
出发菌株为作者所在实验室保藏的C. glutamicum XL-3,由菌株C. glutamicum XQ-9 诱变而来的一株L-亮氨酸生产菌株[19]。 本研究中利用质粒pET-28a 进行蛋白质表达, 利用质粒pK18mobsacB 进行基因的敲除或替换, 大肠杆菌JM109 用于质粒构建,大肠杆菌BL21(DE3)用于表达目的蛋白质。 本实验中所用的主要菌株与质粒如表1 所示,所用引物如表2 所示。
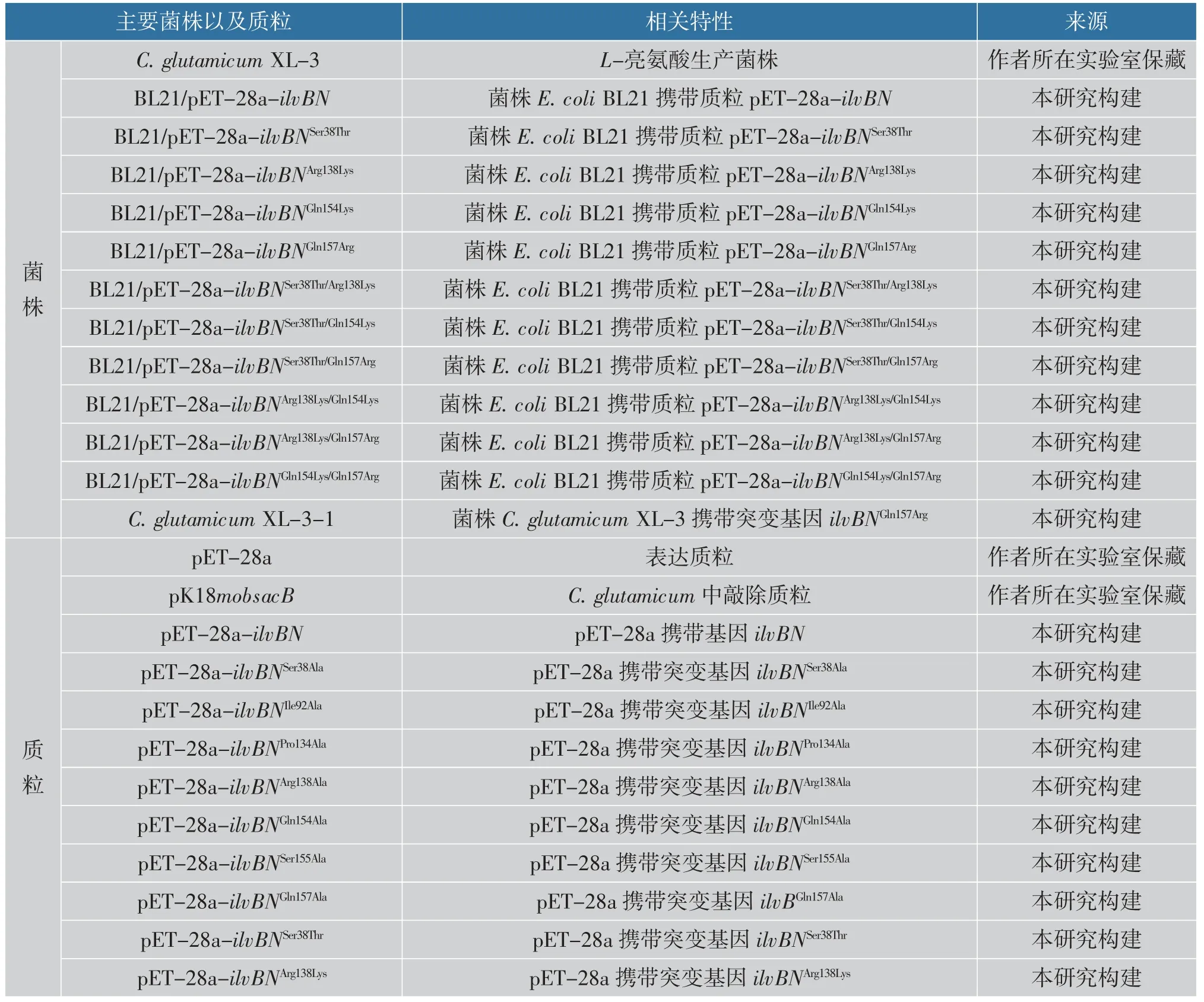
表1 本研究中用到的菌株和质粒Table 1 Strains and plasmids used in this study
1.3 培养基与诱导表达
LB 培养基(g/L):胰蛋白胨10、酵母提取物5、NaCl 10。 LBG 培养基:葡萄糖5、胰蛋白胨10、酵母提取物5、NaCl 10。 EPO 培养基:在LBG 培养基的基础上添加30 g/L 甘氨酸、4 g/L 异烟肼和1 g/L 吐温-80。 LBHIS 培养基:在LB 培养基的基础上添加91 g/L 山梨醇和18.5 g/L 脑心浸出液。 种子培养基(g/L):葡萄糖30、玉米浆30、硫酸铵5、酵母浸膏10、 柠檬酸钠10、K2HPO4·3H2O 1.3、MnSO4·4H2O 0.01、MgSO4·7H2O 0.4、生物素2×10-4、硫胺素3×10-4和L-蛋氨酸0.4。 发酵培养基(g/L):葡萄糖130、玉米浆25、醋酸铵15、硫酸铵15、柠檬酸钠2、MgSO4·7H2O 0.5、K2HPO4·3H2O 1.3、L-蛋氨酸0.7、L-异亮氨酸0.06、L-谷氨酸0.5、MnSO4·H2O 0.01、 生物素1×10-4、硫胺素2×10-4、CaCO330。
大肠杆菌在LB 培养基中生长,培养条件是37 ℃、100 r/min。 谷氨酸棒杆菌在LBG 培养基中生长,培养条件是30 ℃、100 r/min[20]。 使用EPO 培养基和LBHIS 培养基构建谷氨酸棒杆菌重组菌株[21]。此外,用50 μg/mL 卡那霉素构建质粒,用25 μg/mL 卡那霉素筛选重组菌株。
诱导表达:将大肠杆菌BL21(DE3)接种于10 mL 液体LB 培养基中, 在37 ℃、100 r/min 的摇床中培养10~12 h,然后以5%接种体积分数转移到TB 培养基中。当菌液OD600nm达到0.5~0.6 时加入诱导剂IPTG(0.5 mmol/L),然后放置于16 ℃、100 r/min的摇床中培养16~20 h。 将诱导表达培养后的菌体在4 ℃下以10 000 r/min 离心10 min, 用体积分数2%的冷KCl 溶液洗涤细胞2 次。 然后用100 mmol/L磷酸钾缓冲液(pH 7.4)悬浮细胞。 利用超声破碎仪进行细胞破碎15~20 min,然后离心收集上清液,则为粗酶液[11]。
1.4 蛋白质纯化
首先将粗酶液通过0.45 μm 孔径的过滤头进行过滤处理, 然后用结合液 (10 mmol/L 咪唑、100 mmol/L 磷酸钾缓冲液,pH 7.4,15 mL)清洗纯化柱。用洗脱液 (300 mmol/L 咪唑、100 mmol/L 磷酸钾缓冲液,pH 7.4,15 mL)洗脱挂柱的蛋白质。 洗脱时间为20 min, 洗脱过程中结合液和洗脱液体积比从90∶10 至40∶60。纯化后的酶在4 ℃或-80 ℃下保存。
1.5 分析方法
1.5.1 软件分析 本研究中使用了蛋白质分析软件 Schordinger、PyMOL 以及同源建模网站Swissmodel。
1.5.2 目的产物测定 目的产物以及支链氨基酸主要通过高效液相色谱测定[22]。色谱柱为Agilent HC-C18(4.6 mm×250 mm,5 μm),流动相A 为乙腈-水溶液 (体积比60∶40), 流动相B 为乙腈-0.014 mol/L 磷酸盐缓冲液(pH 8.2,体积比20∶80),流量为1.0 mL/min,柱温为30 ℃,检测波长为360 nm,进样体积为10 μL,每个样品30 min,最终流动相A 与B的体积比为80∶20。
1.5.3 酶活力测定 反应体系为磷酸钾缓冲液(100 mmol/L,pH 7.4),含50 mmol/L 丙酮酸钠、10 mmol/L MgCl2、100 mmol/L 硫胺素焦磷酸盐(TPP)和100 mmol/L 黄素腺嘌呤二核苷酸。 首先添加100 μL 酶液至2.5 mg 磷酸钾缓冲液中开始反应,然后在37 ℃下孵育1 h,最后添加200 μL 3 mol/L H2SO4终止反应, 混合物在60 ℃处理15 min。1 mL 体积分数0.5%肌酸和1 mL 体积分数5%α-萘酚(在2.5 mol/L NaOH 中进行制备)添加到混合物中, 然后在60 ℃处理20 min, 最终在525 nm处测量反应的吸光度。 对照组以同样的方式进行处理。 一个酶活力单位被定义为每毫克蛋白质每分钟形成的1 nmol 乙酰乳酸[11]。
1.6 谷氨酸棒杆菌双交换同源重组策略
谷氨酸棒杆菌双交换同源重组策略如图2 所示[23],整个过程使用自杀型质粒pK18mobsacB。首先通过融合PCR 技术将要敲除或替换基因的同源臂进行融合,获得目的片段,然后将目的片段通过酶切酶连技术与空质粒载体pK18mobsacB 连接在一起,获得敲除或替换质粒。 将质粒电转入提前制备好的谷氨酸棒杆菌感受态中完成第一次同源重组,这一步骤通过卡那霉素抗性以及琼脂糖凝胶电泳进行筛选,成功获得完成一次同源的菌株之后进行二次同源筛选。由于质粒pK18mobsacB 携带了sacB基因,此基因编码的蛋白质使得完成一次同源的菌株不能在含有蔗糖的培养基中生存,利用sacB 蔗糖致死原理将质粒从菌株中脱落来完成二次同源,从而获得目的重组菌株。
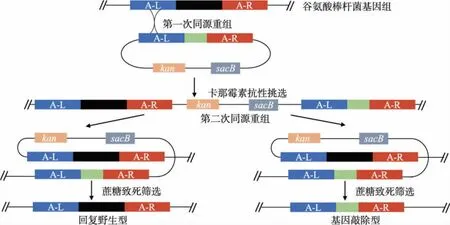
图2 pK18mobsacB 敲除基因流程Fig. 2 Gene knockout of pK18mobsacB plasmid
2 结果与分析
2.1 突变位点的选择
AHAS 是由基因ilvBN 编码的合成支链氨基酸的第一个限速酶,是由两个大亚基和两个小亚基组成的四聚体,效应物主要与小亚基结合,使小亚基结构改变达到催化底物的作用,所以后续的蛋白质改造主要针对AHAS 的小亚基结构 (编码基因ilvN)进行突变改造。代谢过程中丙酮酸与小亚基结合, 催化两分子丙酮酸脱羧缩合形成2-乙酰乳酸,同时也可催化一分子丙酮酸与一分子2-酮基丁酸缩合形成2-乙酰-2-羟基丁酸。当丙酮酸和2-酮基丁酸同时存在时,AHAS 会优先利用2-酮基丁酸合成L-异亮氨酸, 而为了使L-亮氨酸的产量进一步得到优化,本研究中尝试突变AHAS 使其更偏向催化丙酮酸生成2-乙酰乳酸,用于合成目的产物。 考虑到想要改变酶对底物丙酮酸的亲和性,首先增加酶与底物的结合稳定性,所以研究过程中主要针对AHAS 与丙酮酸底物结合位点4 Å 范围内的氨基酸残基进行突变。
目前还未有报道谷氨酸棒杆菌中AHAS 小亚基精确的蛋白质结构,所以首先利用了同源建模网站Swissmodel 进行AHAS 蛋白质结构的预测,以2pc6A(2.5 Å)作为模板同源建模得到了AHAS 小亚基的蛋白质结构。通过分子对接软件Schordinger 将小亚基和丙酮酸进行对接, 然后将对接结构导入PyMOL 进行分析[24]。 结果如图3(a)所示,对接之后发现Ser38、Ile92、Pro134、Arg138、Gln154、Ser155 和Gln157 位于丙酮酸结合位点附近,推测这7 个氨基酸残基可能对底物丙酮酸的结合有一定的作用。 为了找出对底物结合有重要作用的氨基酸残基,分别将以上7 个氨基酸突变成丙氨酸,并测定突变酶催化丙酮酸的酶活力。
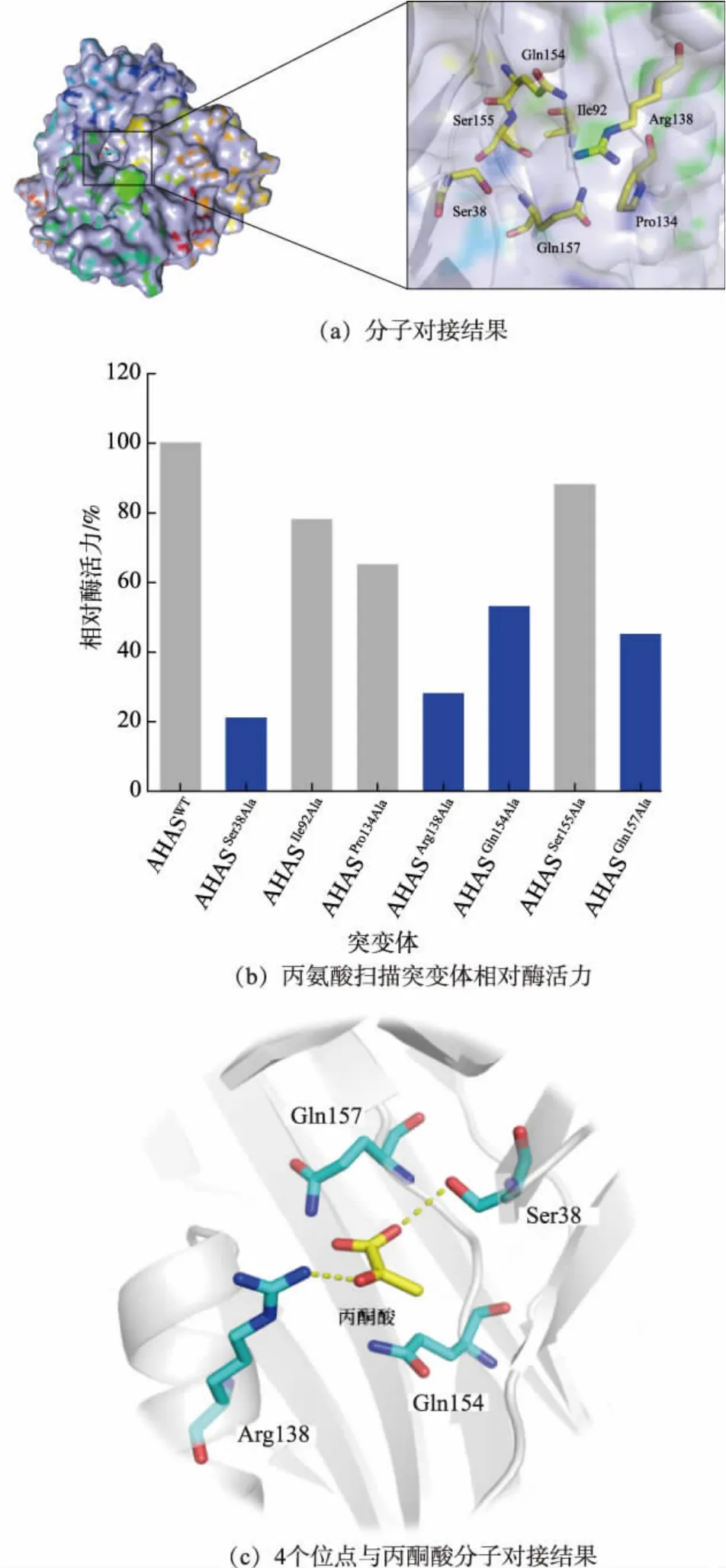
图3 分子对接以及突变体的酶活力分析结果Fig. 3 Results of molecular docking and enzyme activity analysis of mutants
突变体酶活力测定结果如图3(b)所示,可以看出,对比AHASWT(野生型),突变体AHASIle92Ala、AHASPro134Ala以及AHASSer155Ala的酶活力变化较小,推测可能不是催化过程中关键的氨基酸残基,而突变体AHASSer38Ala、AHASArg138Ala、AHASGln154Ala和AHASGln157Ala的酶活力明显下降, 分别下降至初始酶活力的21%、28%、53%和45%, 在一定程度上说明这4 个位点可能对丙酮酸底物结合口袋比较重要。 通过分子模拟软件进一步分析以上位点与丙酮酸的相互作用,结果如图3(c)所示,Ser38、Arg138 与底物丙酮酸之间形成氢键,理论上这两个氨基酸残基对于酶催化底物起着关键的作用,同时这也可能是导致突变体AHASSer38Ala和AHASArg138Ala酶活力明显下降的原因。 Gln154 和Gln157 虽然与丙酮酸没有直接的相互作用力, 但是其突变成Ala 后酶活力也受到了较大的影响,推测这两个位点可能对催化反应有重要的作用。 因此后续研究将围绕这4 个位点进行定点突变来提高酶活力。
2.2 突变体筛选
目前对于蛋白质改造主要通过序列比对、分子模拟技术以及定点突变挖掘潜在保守位点,将氨基酸突变成性质相同或者相反的氨基酸,可能使蛋白质与分子间形成新的空间构象或者形成新的键,从而导致两者结合更加紧密,也可能使底物结合口袋发生变化使得蛋白质对底物的偏好性发生改变。 为了进一步对Ser38、Arg138、Gln154 和Gln157 这4个位点进行研究, 尝试进行以下突变Ser38Thr、Arg138Lys、Gln154Lys 以及Gln157Arg,通过定点突变试剂盒得到突变质粒,将质粒转化到菌株大肠杆菌BL21(DE3)中获得重组菌株BL21/pET-28ailvBNSer38Thr、BL21/pET -28a -ilvBNArg138Lys、BL21/pET -28a-ilvBNGln154Lys和BL21/pET-28a-ilvBNGln157Arg, 酶活力测定方法参考1.5.3 部分。
单突变体酶活力测定结果如图4 (a) 所示,与AHASWT相比较,AHASArg138Lys和AHASGln154Lys分别下降了13%和2%, 但是AHASSer38Thr和AHASGln157Arg的酶活力分别上升了14%和41%,结果表明通过单突变使得酶活力有了一定的提升。 为了进一步提高酶活力, 将选择的4 种突变体分别进行两两组合突变, 从而获得了新的突变体AHASSer38Thr/Arg138Lys、AHASSer38Thr/Gln154Lys、AHASSer38Thr/Gln157Arg、AHASArg138Lys/Gln154Lys、AHASArg138Lys/Gln157Arg以及AHASGln154Lys/Gln157Arg。双突变体酶活力测定结果如图4(b)所示,突变体AHASSer38Thr/Gln154Lys和AHASArg138Lys/Gln157Arg的酶活力与AHASWT相比分别上升了20%和14%, 但是都没有单突变体AHASGln157Arg酶活力高,推测可能是组合突变对酶的催化空间构型产生了较大的影响,使得蛋白质与底物结合不紧密。 实验中测定了野生型和突变体酶对丙酮酸的动力学参数,结果如表3 所示,与AHASWT相比, 突变体AHASGln157Arg对底物丙酮酸的Km值下降,说明将AHAS 的157 位从Gln 突变为Arg 后在一定程度上改变了底物偏好性,提高了酶对丙酮酸的催化能力。 同时从表中可以得到突变体AHASGln157Arg的Kcat/Km高于AHASWT, 所以突变酶与野生酶相比有更高的催化效率, 最终选择AHASGln157Arg为最优突变体。
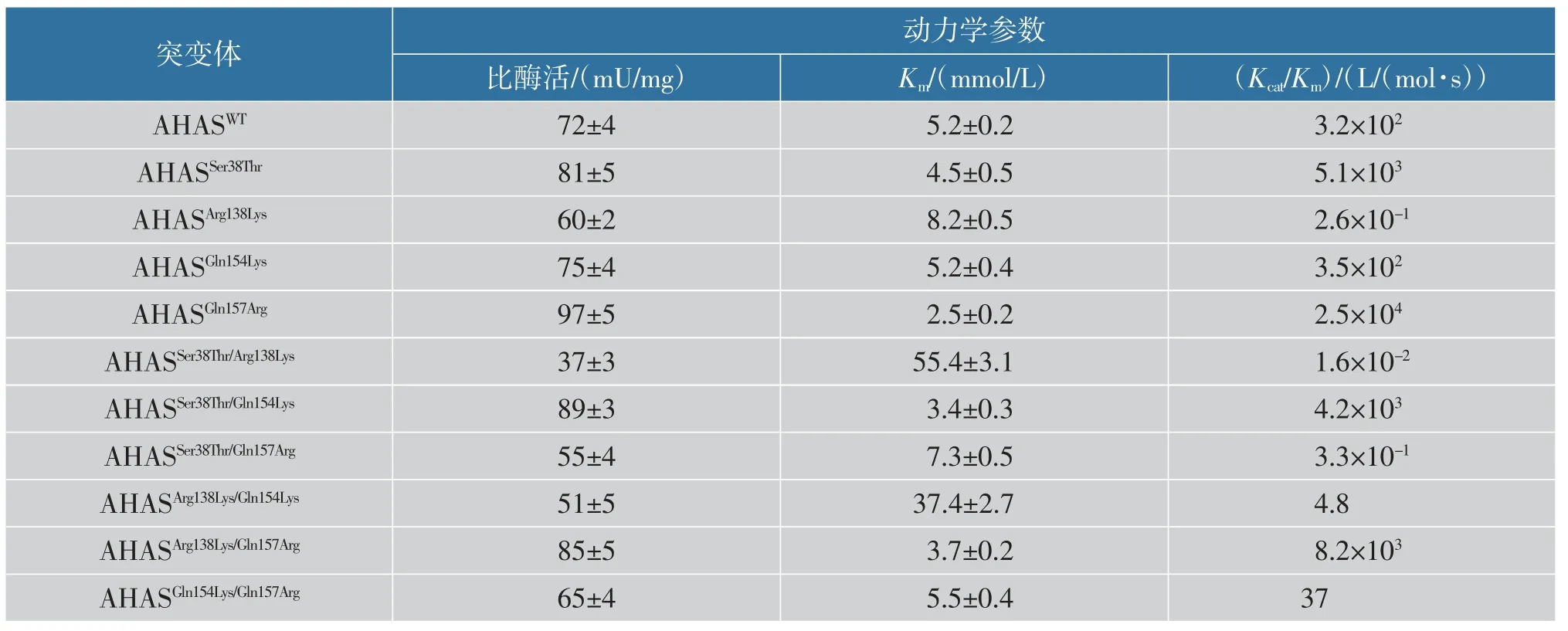
表3 野生型和突变体酶对丙酮酸合成的动力学参数Table 3 Kinetic parameters of wild-type enzyme and mutants to pyruvate
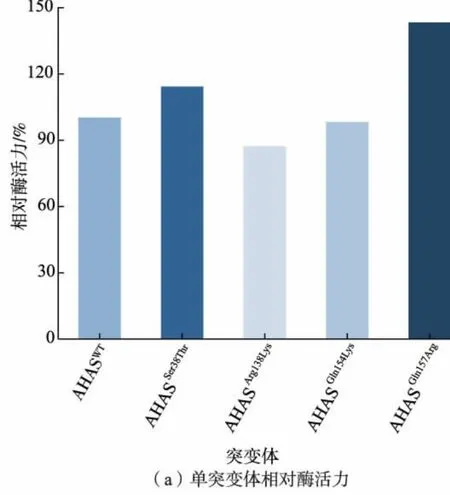
图4 突变体的酶活力分析Fig. 4 Enzyme activity analysis of mutants
2.3 AHAS 及突变体结构分析
通过上述实验得到了一个最佳的突变体AHASGln157Arg,提高了酶活力,且显著提高了AHAS 对底物丙酮酸的亲和性。 为了进一步探究酶活力提高的原因, 利用蛋白质分子软件对突变体进行解析。首先利用分子对接软件Schordinger 将突变后的酶与底物丙酮酸和2-酮基丁酸进行对接,随后将结果导入PyMOL 进行相互作用的分析[24],分析结果如图5 所示。 由图5(a)可知,AHAS 的Gln157 在没有突变之前虽然与底物丙酮酸结合位点比较相近,但是与丙酮酸没有直接的相互作用。 然而突变体AHASGln157Arg的氨基酸残基Arg157 与底物丙酮酸之间多了一个氢键(见图5(b)),氢键对于蛋白质与小分子配体之间起着十分关键的作用,这使得丙酮酸与AHAS 之间结合更加稳定, 增加了底物亲和性,这可能是突变提高酶活力的重要原因。其次,157 位氨基酸从Gln 突变为Arg 后氨基酸残基的侧链变大,这个改变可能会缩小底物结合口袋,而丙酮酸体积本身就比较小, 缩小的口袋就会更契合丙酮酸,推测这也是突变体酶活力提高的原因之一。
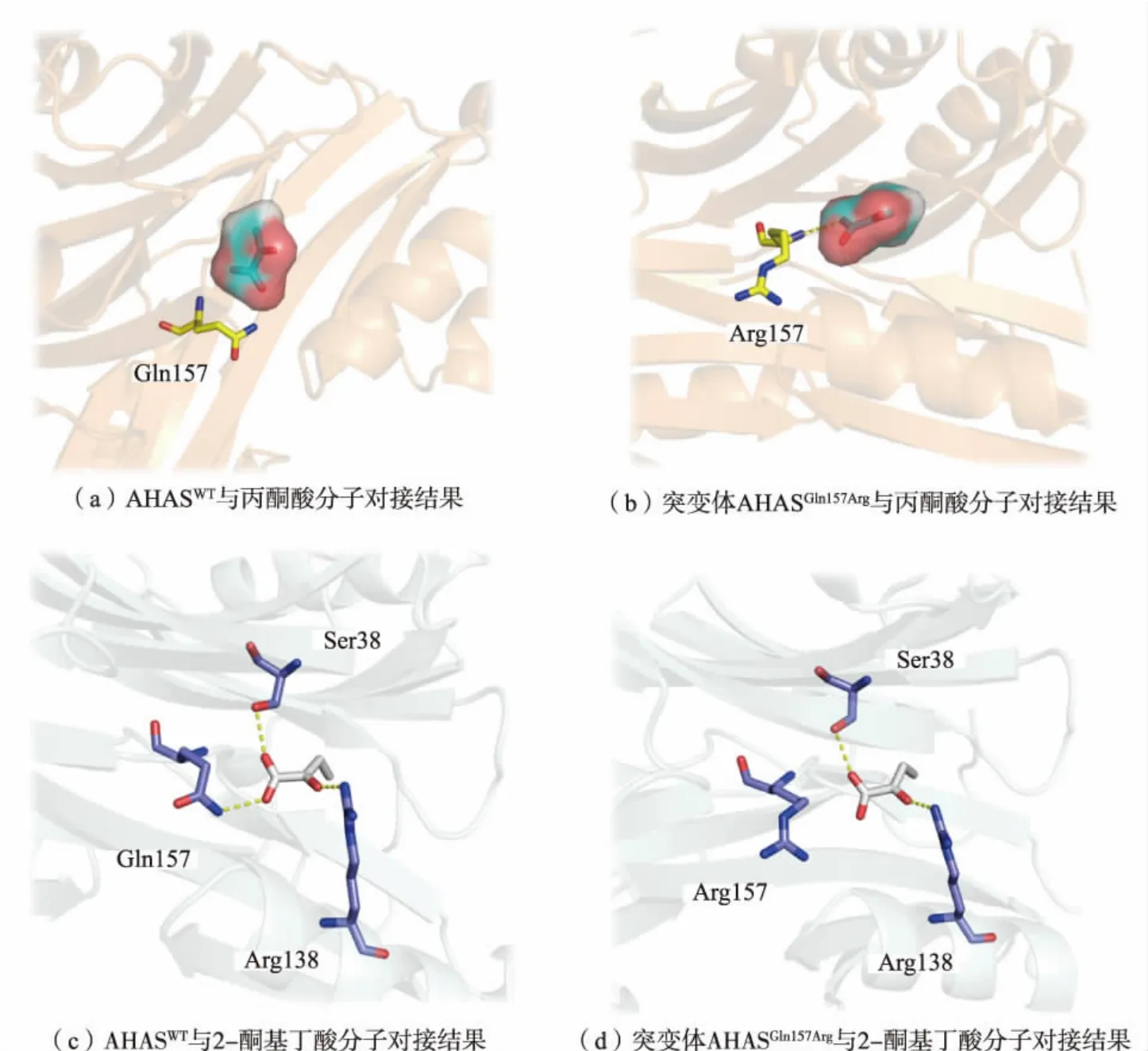
图5 突变前后蛋白质与底物之间的相互作用Fig. 5 Interaction between protein and substrate before and after mutation
此外,分析了AHASWT和AHASGln157Arg与底物2-酮基丁酸的结合情况。如图5(c)所示,AHASWT与底物2-酮基丁酸之间形成了3 个氢键,氨基酸结合位点分别是Ser38、Gln157 以及Arg138。当157 位氨基酸从Gln 突变为Arg 后, 突变体AHASGln157Arg的157位氨基酸残基失去了与2-酮基丁酸的氢键作用(见图5(d)),这可能会减弱突变体对2-酮基丁酸的亲和力, 改变其对底物丙酮酸和2-酮基丁酸的偏好性, 从而使更多的碳通量流向L-亮氨酸合成途径,减少副产物L-异亮氨酸的积累。
2.4 重组菌株的产量测定
选择了最优突变体AHASGln157Arg,使用谷氨酸棒杆菌双交换同源重组策略将突变位点引入到基因组,菌株构建过程参考1.6 部分。 通过测序验证获得重组菌株C. glutamicum XL-3-ilvBNGln157Arg(C. glutamicum XL-3-1)。 将获得的重组菌株C. glutamicum XL-3-1 摇瓶发酵结果进行测定,结果如图6 所示。 从图中可以得出重组菌株C. glutamicum XL-3-1 与出发菌株的生长情况相近,说明引入的突变并未对菌株的生长造成影响,其L-亮氨酸产量达到了(23.5±1.8)g/L,与出发菌株相比产量提高了51%;副产物L-缬氨酸产量为(4.7±0.7)g/L,与出发菌株相比产量提高了18%;副产物L-异亮氨酸产量为(1.2±0.3) g/L,与出发菌株相比产量下降了45%。 从产量结果表明,将AHAS 的Gln157 位点突变成Arg 能够有效提高AHAS 催化丙酮酸的能力, 减弱其对2-酮基丁酸的亲和力, 实现了AHAS底物偏好性的改变,在提高L-亮氨酸产量的同时减少了副产物L-异亮氨酸的积累。
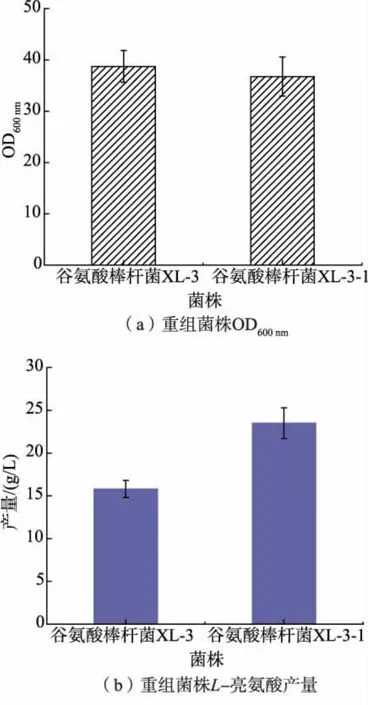
图6 摇瓶发酵产物分析Fig. 6 Analysis of shake flask fermentation products
3 结 语
目前关于支链氨基酸主要集中在蛋白质水平上解除支链氨基酸对AHAS 的反馈抑制以及改造代谢途径减少副产物来实现支链氨基酸的积累[25-26]。然而,关于通过蛋白质改造改变AHAS 的底物偏好性增加支链氨基酸积累的研究较少。 本研究中围绕谷氨酸棒杆菌中AHAS 的调节亚基进行定点突变,提高了酶催化底物丙酮酸的活性,从而提高丙酮酸流向L-亮氨酸的代谢通量。首先通过同源建模得到了小亚基的蛋白质结构,然后利用Schordinger 通过分子对接找到AHAS 与丙酮酸底物结合位点4 Å范围内的7 个氨基酸残基, 分别为Ser38、Ile92、Pro134、Arg138、Gln154、Ser155 和Gln157, 之后通过丙氨酸扫描确定了4 个重要的氨基酸残基(Ser38、Arg138、Gln154 和Gln157)。通过测定单突变体 AHASSer38Thr、AHASArg138Lys、AHASGln154Lys、AHASGln157Arg以及双突变体AHASSer38Thr/Arg138Lys、AHASSer38Thr/Gln154Lys、AHASSer38Thr/Gln157Arg、AHASArg138Lys/Gln154Lys、AHASArg138Lys/Gln157Arg、AHASGln154Lys/Gln157Arg酶活力,发现单突变AHASGln157Arg的酶活力最高,比AHASWT提高了41%。通过软件分析可知,酶活力提高可能在于底物与酶结合更稳定或者底物口袋缩小增加了底物亲和性。 最后将最优突变体AHASGln157Arg引入,获得重组菌株谷氨酸棒杆菌XL-3-1。 对重组菌株进行摇瓶发酵可知,L-亮氨酸产量为(23.5±1.8) g/L,与出发菌株相比产量提高了51%。本研究为AHAS 蛋白质改造促进L-亮氨酸的生物合成提供了一种新的思路。
猜你喜欢
工业微生物(2024年1期)2024-02-29 07:36:50
生物加工过程(2023年6期)2023-12-11 03:27:52
牡丹江医学院学报(2021年5期)2021-12-05 08:01:51
中国饲料(2021年17期)2021-11-02 08:15:10
当代水产(2021年2期)2021-03-29 02:57:48
传染病信息(2021年6期)2021-02-12 01:52:14
烟草科技(2015年8期)2015-12-20 08:27:06
动物营养学报(2015年10期)2015-12-01 02:26:21
大连工业大学学报(2014年2期)2014-09-19 08:52:52
食品科学(2013年19期)2013-03-11 18:27:35
