
超高效液相色谱指纹图谱法和一测多评法评价不同产地西红花质量*
2023-10-17 06:23张梦奇王琼芬张红萍郑国平
中国药业 2023年19期
张梦奇,王琼芬,李 彬,刘 婷,张红萍,石 婧,徐 虹,郑国平
(浙江省舟山市食品药品检验检测研究院,浙江舟山 316000)
西红花为鸢尾科植物番红花Crocus sativusL. 的干燥柱头,原产于地中海、欧洲南部地区和伊朗,现我国多个省份均有栽培。西红花不仅是世界著名的天然香料和食用染料,也是名贵的中药材,收载于2020 年版《中国药典(一部)》,具有活血化瘀、凉血解毒、解郁安神等功效[1-2]。其发挥药效的主要物质基础为类胡萝卜素及其糖苷类衍生物,包括西红花苷类(西红花苷-Ⅰ、西红花苷- Ⅱ、西红花苷- Ⅲ和西红花苷- Ⅳ)、苦番红花素、西红花酸和西红花醛[3-4]。有关西红花的研究主要集中于指标成分定量测定[5],也有用于质量评价的指纹图谱研究[6-7],但尚未见结合化学模式识别用于产地区分的相关报道。指纹图谱结合化学模式识别分析可有效表征和评价中药的内在和整体质量,并解释药材复杂成分的隐藏规律,客观评价不同产地间的品质差异[8]。西红花相关对照品主要提取于西红花药材[9],检测成本高。一测多评(QAMS)是适合中药特点的多指标成分质量评价新模式,在中药多指标成分的含量测定中应用广泛[10],可大幅降低检测成本。故本研究中建立了西红花的超高效液相色谱(UPLC)指纹图谱,采用相似度评价、聚类分析和正交偏最小二乘法- 判别分析(OPLS-DA)对市售西红花药材的质量进行综合评价;并建立了以西红花苷- Ⅰ作为内参物,测定苦番红花素及4 种西红花苷类成分的QAMS 法,旨在为不同产地西红花药材的质量控制和评价提供参考。现报道如下。
1 仪器与试药
1.1 仪器
Agilent 1260 Infinity Ⅱ型高效液相色谱仪(美国Agilent 公司);Waters Arc 型超高效液相色谱仪(美国Waters 公司);Shimadzu LC-30A 型超高效液相色谱仪(日本Shimadzu公司);XSE205DU型电子天平(梅特勒-托利多仪器<上海>有限公司,精度为0.01 mg);KQ-300VDE 型双频数控超声波清洗器(昆山市超声仪器有限公司,功率为300 W,频率为45 kHz);Milli - Q 型超纯水仪(美国Milipore公司)。
1.2 试药
苦番红花素对照品(批号为112056-202102,含量为97.3%),西红花苷- Ⅰ对照品(批号为111588 -201704,含量为88.4%),西红花苷-Ⅱ对照品(批号为111589-201705,含量为92.2%),均购自中国食品药品检定研究院;西红花苷-Ⅲ对照品(四川省维克奇生物科技有限公司,批号为wkq-21081003,含量为99.75%);西红花苷-Ⅳ对照品(成都乐美天医药科技有限公司,批号为DST201110-181,含量为98.07%);甲醇、乙腈(色谱纯,J. T. Baker 公司);水为Milli-Q自制水,其他试剂均为分析纯;30批西红花药材样品均购自浙江省内药品批发、零售企业,经浙江省舟山市食品药品检验检测研究院郑国平主任中药师鉴定为正品,样品信息见表1。

表1 30批西红花药材样品信息Tab.1 Information of 30 batches of Croci Stigma
2 方法与结果
2.1 色谱条件
色谱柱:Waters Acquity UPLC HSS T3 柱(100 mm×2.1 mm,1.8µm);流动相:乙腈(A)-水(B),梯度洗脱(0~6 min 时15%A →30%A,6~12 min 时30%A,12~14 min 时30%A →35%A,14~16 min 时35%A →50%A,16~20 min 时50%A →60%A,20~25 min 时60%A);流速:0.3 mL/min;检测波长:254 nm(0~8 min,苦番红花素),440 nm(8~25 min,西红花苷-Ⅰ、西红花苷- Ⅱ、西红花苷- Ⅲ和西红花苷- Ⅳ);柱温:25 ℃;进样量:4µL。
2.2 溶液制备
分别取苦番红花素、西红花苷-Ⅰ、西红花苷-Ⅱ、西红花苷- Ⅲ和西红花苷- Ⅳ对照品各适量,精密称定,加稀乙醇制成质量浓度分别为18.27,28.85,9.95,2.96,1.18 µg/ mL 的混合对照品溶液。取西红花药材细粉10 mg,精密称定,置50 mL 棕色容量瓶中,加稀乙醇适量,置冰浴中超声处理20 min,放至室温,加稀乙醇定容,摇匀,滤过,取续滤液,即得供试品溶液。
2.3 UPLC 指纹图谱建立与分析
2.3.1 方法学考察
精密度试验:精密量取2.2 项下供试品溶液适量,按2.1 项下色谱条件连续进样测定6 次,以峰4 为参照峰,分别计算各共有峰的相对峰面积和相对保留时间。结果的RSD分别为0.19%~1.94%和0.06%~0.24%(n=6),表明方法精密度良好。
稳定性试验:取样品(编号S16),按2.2项下方法制备供试品溶液,分别于室温下放置0,2,4,8,10,12 h 时按2.1 项下色谱条件进样测定,以峰4 为参照峰,分别计算各共有峰的相对峰面积和相对保留时间。结果的RSD分别为0.21%~1.84%和0.03%~0.37%(n=6),表明供试品溶液在室温下放置12 h内稳定性良好。
重复性试验:取样品(编号S16),取稳定性试验项下供试品溶液6 份,按2.1 项下色谱条件进样测定,以峰4为参照峰,计算各共有峰的相对峰面积和相对保留时间。结果的RSD分别为0.17%~2.23%和0.04%~0.95%(n=6),表明方法重复性良好。
2.3.2 指纹图谱生成与相似度评价
取30批(编号为S1-S30)西红花药材各适量,精密称定,按2.2 项下方法制备供试品溶液,按2.1 项下色谱条件进样测定,记录色谱图。采用中药色谱指纹图谱相似度评价系统(2012 版)对色谱图进行分析,将S1 样品的图谱作为参照图谱,时间窗为0.1 min,采用中位数法,经过多点校正和全谱峰匹配,生成对照指纹图谱(R),共标识11 个共有峰,详见图1。通过与混合对照品溶液色谱图进行比对,指认出5 个共有峰,其中,峰1 为苦番红花素,峰4为西红花苷-Ⅰ(参照峰S),峰6为西红花苷-Ⅱ,峰7为西红花苷-Ⅳ,峰10为西红花苷-Ⅲ,详见图2。30 批样品指纹图谱相似度为0.983~0.999,详见表2。所有样品相似度均大于0.98,表明不同产地、不同批次西红花药材所含成分基本一致,药材整体质量较稳定,所建立的指纹图谱可用于西红花药材的质量评价。
目前,三方共同创新研制了名为“智宝”的移动式病虫害智能化感知设备,填补了国内外移动式病虫害智能测报技术与产品的空白。该设备集成了包括视觉、温湿度传感器、地理位置、移动终端等多种信息获取手段,有效提升了现有病虫害监测能力。

图1 30批西红花药材超高效液相色谱叠加指纹图谱和对照指纹图谱Fig.1 UPLC overlay fingerprints of 30 batches of Croci Stigma and the reference fingerprint

图2 超高效液相色谱图1.Picrocrocin 4.Crocin I 6.Crocin Ⅱ 7.Crocin Ⅳ 10.Crocin ⅢA.Mixed reference solution B.Test solutionFig.2 UPLC chromatograms
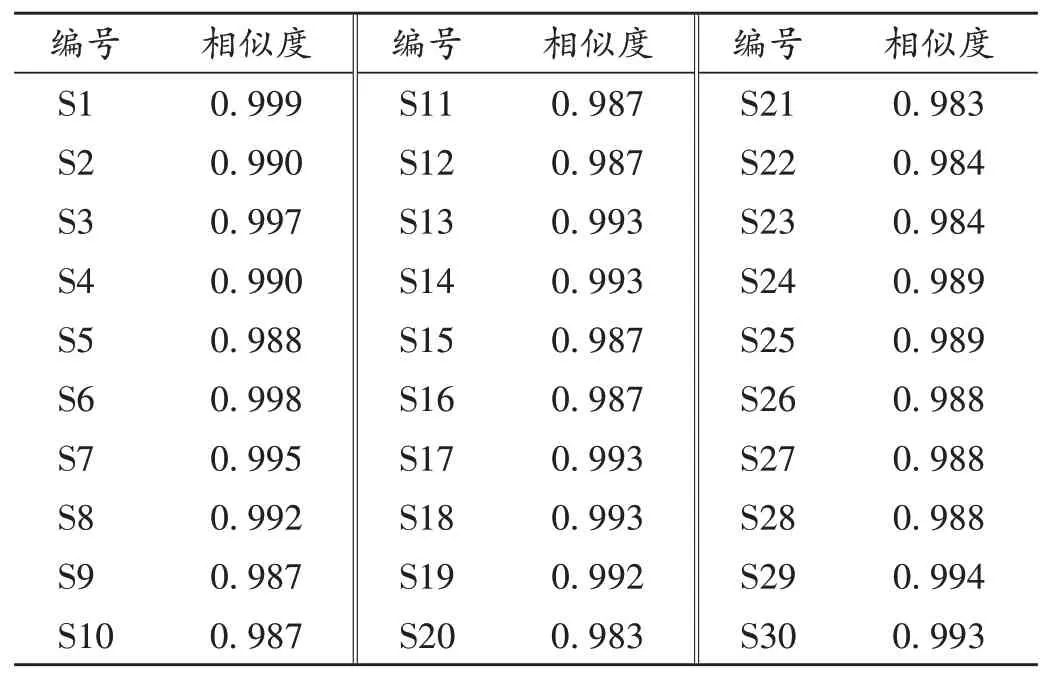
表2 30批西红花药材指纹图谱相似度评价结果Tab.2 Results of the similarity evaluation of fingerprints of 30 batches of Croci Stigma
2.4 聚类分析
以30 批西红花药材的11 个共有峰的峰面积为变量,导入SPSS 26.0 统计学软件,采用组间联接,以平方欧氏距离法进行系统聚类分析,并绘制谱系图,结果见图3。当欧氏距离为20 时,30 批西红花药材样品可聚为两大类,其中,编号为S1,S2,S3(产地伊朗)的样品聚为一类,剩余批次聚为二类(国产)。当欧氏距离为5 时,30 批西红花药材样品可聚为4 类。其中,一类为编号为S1-S3(产地伊朗)的样品;二类为编号为S4-S6(产地西藏),S8,S10,S12,S13(产地浙江),S22(产地江苏)的样品;三类为编号为S25-S30(产地安徽)的样品;其余批次为四类(产地浙江、江苏)。由聚类分析结果可知,产地为伊朗的西红花与产地为我国的差异明显,而我国不同产地之间也存在一定差异,但产地为浙江、西藏、江苏的药材质量较相似,可用聚类分析法对药材样品进行产地区分。

图3 30批西红花药材的聚类分析树状图Fig.3 Cluster analysis diagram of 30 batches of Croci Stigma
2.5 OPLS-DA
将30 批西红花药材的11 个共有峰的峰面积作为变量,导入SIMCA 14.1软件进行OPLS-DA 分析,获得相应模型,得分图见图4。可见,30批西红花药材样品聚为4 类,编号为S1-S3 的样品聚为一类(产地伊朗)、编号为S4-S6的样品聚为二类(产地西藏)、编号为S25-S30的样品聚为三类(产地安徽)编号为S7-S24的样品聚为四类(产地浙江、江苏),各组样品聚类良好、分离明显。模型的自变量模型参数R2X(cum)为0.809(>0.5),因变量模型参数R2Y(cum)为0.800(>0.5),表明模型稳定性及预测力较好;累积预测能力参数Q2(cum)为0.751 >0.5),且R2Y(cum)-Q2(cum)= 0.049 <0.3。表明预测模型具有较好的准确性和可靠性[11]。

图4 30批西红花药材的OPLS-DA模型得分图Fig.4 OPLS-DA scores diagram of 30 batches of Croci Stigma
为进一步检验模型的有效性或样本间是否存在差异,利用SIMCA 14.1 软件对已建立的OPLS - DA 模型进行置换检验,设置迭代次数为200 次,结果见图5。可见,代表模型解释能力R2为(0,0.064 5),模型预测能力Q2为(0,-0.38),均小于原始值,表明所建模型未过拟合[12],可用于有效区分不同产地的西红花药材。

图5 OPLS-DA模型置换检验图Fig.5 OPLS-DA model permutation test diagram
变量重要性投影(VIP)值可衡量各共有峰的表达模式对样本分类判别的影响强度和解释能力[13],且当VIP >1.0 时,表示该变量对于所建模型的贡献度高于平均水平[14]。以此为筛选标准,共筛选出5 种贡献较大的成分,依次为峰8、峰9、峰11、峰10(西红花苷-Ⅲ)、峰2。详见图6。

图6 共有峰峰面积的VIP图Fig.6 VIP plot of the common peak area
2.6 含量测定方法学考察
线性关系考察:分别精密量取2.2项下混合对照品溶液0.1,0.2,0.5,1.0,2.0,5.0 mL,分别置10 mL棕色容量瓶中,用稀乙醇定容,摇匀,制成系列混合对照品溶液,按2.1 项下色谱条件依次进样测定,以待测成分质量浓度(X,µg/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归。结果见表3,表明各成分在相应质量浓度范围内与峰面积线性关系良好。

表3 5种成分的线性关系考察与检测限、定量限确定结果Tab.3 Results of the linear relation test,LOD and LOQ of five components
检测限(LOD)与定量限(LOQ)确定:精密吸取2.2项下混合对照品溶液适量,用稀乙醇逐级稀释,按2.1项下色谱条件进样测定,以各仪器信噪比(S/N)为10∶1时的待测成分质量浓度为LOQ,以S/N为3∶1 时的质量浓度为LOD。结果见表3。
精密度试验:精密吸取2.2项下混合对照品溶液适量,按2.1 项下色谱条件连续进样测定6 次,记录峰面积。结果苦番红花素、西红花苷-Ⅰ、西红花苷-Ⅱ、西红花苷- Ⅲ、西红花苷- Ⅳ峰面积的RSD分别为0.17%,0.22%,0.30%,0.51%,0.43%(n= 6),表明仪器精密度良好。
重复性试验:取西红花药材(编号为S16)细粉适量,精密称定,共6 份,按2.2 项下方法制备供试品溶液,按2.1项下色谱条件进样测定,记录峰面积。结果苦番红花素、西红花苷-Ⅰ、西红花苷-Ⅱ、西红花苷-Ⅲ、西红花苷- Ⅳ的RSD分别为0.55%,0.18%,0.15%,0.73%,1.09%(n=6),表明方法重复性良好。
稳定性试验:取重复性试验项下供试品溶液,分别于室温放置0,2,4,8,10,12 h按2.1项下色谱条件进样测定,记录各成分峰面积,并计算含量。结果苦番红花素、西红花苷-Ⅰ、西红花苷-Ⅱ、西红花苷-Ⅲ、西红花苷- Ⅳ含量的RSD分别为1.12%,1.59%,1.63%,1.71%,1.92%(n= 6),表明供试品溶液在12 h 内稳定性良好。
加样回收试验:取西红花药材(编号为S16)细粉10 mg,精密称定,置100 mL 棕色容量瓶中,共6 份,按2.2 项下方法制备质量浓度分别为112.64,99.81,33.24,8.93,3.82 µg/ mL 的混合对照品溶液,精密量取混合对照品溶液8,10,12 mL,分别加入相应供试品溶液中,按2.2 项下方法制备供试品加标溶液,按2.1项下色谱条件进样测定,并计算加样回收率。结果苦番红花素、西红花苷-Ⅰ、西红花苷-Ⅱ、西红花苷-Ⅲ、西红花苷- Ⅳ的平均加样回收率分别为100.47%,99.79%,99.87%,100.27%,99.73%,RSD分别为0.94%,0.89%,1.18%,1.23%,1.74%(n= 6),表明方法准确性良好。
2.7 QAMS 建立
相对校正因子(RCF,fs/i)计算:在中药材多成分指标质量评价时,以该药材中某一代表成分为内参物,通过建立该内参物与其他待测成分的fs/i计算其他待测组分的含量[15],实现中药材多成分同步测定。本研究中采用多点校正法,取线性关系考察项下系列混合对照品溶液,依次进样测定,记录峰面积。以西红花苷- Ⅰ作为内参物,按公式fs/i=(As×Mi)/(Ai×Ms)计算苦番红花素、西红花苷- Ⅱ、西红花苷- Ⅲ、西红花苷- Ⅳ的fs/i及RSD。其中,As和Ai分别为内参物和待测成分的峰面积,Ms和Mi分别为内参物和待测成分对照品的质量浓度。结果fs/i分别为3.577,0.844,1.251,1.781,RSD分别为0.88%,0.82%,1.39%,1.98%(n=6)。
耐用性试验:分别考察不同色谱柱[(Waters Acquity UPLC HSS T3柱(100 mm×2.1 mm,1.8µm),月旭Ultimate UHPLC C18柱(100 mm×2.1 mm,1.8µm),Agilent Poroshell 120 SB-C18柱(100 mm×2.1 mm,1.9µm)],不同品牌色谱仪(Agilent 1260型、Waters Arc型、Shimadzu LC-30A 型),不同流速(0.25,0.30,0.35 mL/min),不同柱温(25,30,35 ℃)对各成分fs/i的影响。结果fs/i的RSD均低于2.0%,表明不同色谱条件对苦番红花素、西红花苷- Ⅱ、西红花苷- Ⅲ、西红花苷- Ⅳ的fs/i均无显著影响。
待测成分色谱峰定位:分别考察了苦番红花素、西红花-Ⅱ、西红花苷-Ⅲ、西红花苷-Ⅳ与西红花苷-Ⅰ间的相对保留时间(Ri/s=ti/ts)和保留时间差(ti-ts)在不同色谱仪和色谱柱中的重复性。结果表明,相对保留时间的波动较小,其重复性明显优于保留时间差,故本研究中选择相对保留时间作为待测成分色谱峰定位依据。
样品含量测定:取30批(编号为S1-S30)样品各适量,精密称定,分别制备供试品溶液,按2.1项下色谱条件进样测定,采用外标法(ESM)测定苦番红花素、西红花苷-Ⅰ、西红花苷-Ⅱ、西红花苷-Ⅲ、西红花苷-Ⅳ的含量;同时采用QAMS 法以西红花苷-Ⅰ为内参物,计算苦番红花素、西红花-Ⅱ、西红花苷-Ⅲ和西红花苷-Ⅳ的含量,并与ESM 法测定结果进行比较,结果见表4。可见,各成分QAMS法计算值和外标实测值间无显著差异,相对偏差(RD)低于2.0%。同时,采用SPSS 22.0统计学软件对2种方法所测得含量进行t检验,结果显示,2种方法无显著差异(P>0.05)。以上述5种成分的总量计,浙江、伊朗产药材含量较高,安徽产药材含量最低。
3 讨论
3.1 色谱条件优化
2020 年版《中国药典(一部)》收载的西红花药材含量测定方法采用高效液相色谱(HPLC)法,梯度洗脱时间约为60 min,检测时间较长。本研究中采用UPLC 法,将检测时间缩短至25 min 内,有效提高了检测效率,还减少了有机溶剂的使用量。同时,采用梯度洗脱,在保留时间为15~25 min 处获得多个分离良好的其他西红花苷成分(西红花苷-Ⅲ、西红花苷-Ⅳ等),但上述成分在常规HPLC 法检测中难以获得良好分离。通过二极管阵列检测器于190~800 nm 波长范围内进行全波长扫描发现,苦番红花素于254 nm 波长处有最大吸收,而其他西红花苷类成分于440 nm 波长处响应较强,故采用波长切换法,以保证检测灵敏度。优化色谱条件后,各成分间分离良好,可满足西红花药材指纹图谱及QAMS法分析的要求。
3.2 指纹图谱建立、聚类分析与OPLS-DA
本研究中建立了西红花药材的UPLC 指纹图谱,共检出11个共有峰。相似度评价结果显示,30批西红花药材相似度为0.983~0.999,表明不同产地西红花药材的化学成分种类差异小。聚类分析和OPLS-DA结果均将30 批西红花药材分为4 类,聚类分析可实现伊朗、安徽和其他产地西红花的有效区分,而OPLS-DA模型可实现伊朗、西藏、安徽产地西红花的有效区分,但仍无法有效区分浙江和江苏2个产地的西红花药材,两者质量高度相近。通过VIP 预测值排序筛选出5 个贡献度较大的成分,分别为峰8、峰9、峰11、峰10(西红花苷-Ⅲ)、峰2,可作为西红花产地区分的标志物。
3.3 QAMS 法测定结果分析
QAMS 法可有效解决对照品不易获得、化学性质不稳定、价格昂贵、溶液制备烦琐等含量测定不利因素。本研究发现,西红花药材中西红花苷-Ⅰ含量较高、化学性质较稳定且价格低,故选用西红花苷- Ⅰ作为内参物。方法学验证、方法耐用性考察,结果显示,西红花苷- Ⅰ对苦番红花素、西红花苷- Ⅱ、西红花苷- Ⅲ、西红花苷-Ⅳ的fs/i有较好的重复性,同时QAMS法计算含量与ESM 法实测含量无显著差异(RD<2.0%),建立的QAMS法适用于西红花中多指标成分的含量测定。含量测定结果显示,不同产地、不同批次西红花苦番红花素和西红花苷类成分总含量差异较大。其中,产地为伊朗和浙江的西红花各成分总含量较高,产地为安徽的西红花药材含量较低,且含量较低样品普遍为生产日期均早、包装材料密封性差的样品。因此,西红花内在品质除与其药材产地相关外,还与储存时间、储存方式相关。
3.4 方法评价
本研究中建立的UPLC 指纹图谱结合QAMS 法准确、可靠、便捷,且检测成本低,可用于西红花药材的质量评价及产地区分。
猜你喜欢
快乐语文(2021年34期)2022-01-18
快乐语文(2021年27期)2021-11-24
快乐语文(2021年11期)2021-07-20
快乐语文(2021年15期)2021-06-15
饮食科学(2019年9期)2019-09-23
中国外汇(2019年22期)2019-05-21
意林·全彩Color(2018年9期)2018-10-12
中成药(2018年8期)2018-08-29
兽医导刊(2016年6期)2016-05-17
中央民族大学学报(自然科学版)(2015年3期)2015-06-11
