
基于加权平均的肠道菌群特征筛选和疾病预测模型研究
2023-10-14 02:14曹海涛朱静曾海波刘彦辰
生物技术进展 2023年5期
曹海涛 , 朱静 , 曾海波 , 刘彦辰
1.新疆农业大学计算机与信息工程学院,乌鲁木齐 830052;2.新疆乌鲁木齐市友谊医院,乌鲁木齐 830049
传统疾病筛查和诊断通常检测周期较长,且一些筛查和诊断方法需要进行侵入性检测,例如穿刺、活组织检查等。这种检测方式不仅痛苦,还可能导致感染和其他并发症,使患者出现紧张、不适[1]等情绪。因此,需要寻找一种更便捷、非侵入性的方法来辅助疾病筛查和诊断,以改善早期诊断和治疗效果,改善人们健康状况。本研究旨在利用宏基因组学和机器学习技术,探索肠道菌群[2]在疾病筛查和诊断中的应用,以建立一种辅助参考模型。
近年来,肠道菌群对疾病的影响引起了广泛关注。研究表明,患有某些疾病的人群其肠道菌群与健康人群存在差异[3],肠道微生物群落的失衡可能导致病原菌过度生长和有害代谢产物的产生,从而引起免疫系统异常和疾病的发生,如克罗恩病[4]和糖尿病[5]等。然而,现有的疾病诊断[6]方法存在局限性,单一模型的诊断能力有限,而利用多组数据建立模型需要大量的特征,难以应用于临床。1998年,Handelsman等[7]首次提出宏基因组学(metagenomics)的概念——一种研究环境中所有微生物基因组总体的方法,高通量测序技术的发展[8]极大地推动了宏基因组学的研究。宏基因组学为我们更好地理解和利用肠道菌群提供了平台,从而深入研究人体肠道菌群与健康之间的关系。
机器学习通常作为各种预测任务模型的核心算法使用,在构建疾病预测模型时,大量使用逻辑回归、K近邻、随机森林和人工神经网络等方法。Pasolli等[9]使用随机森林模型设计了有关2型糖尿病的疾病预测模型;Ai等[10]使用随机森林完善了关于结直肠癌的疾病预测模型;Wu等[11]使用K近邻建立了预测2型糖尿病的疾病模型;Reiman等[12]使用人工神经网络构建了预测肝硬化疾病模型。尽管使用不同模型预测疾病的方法大部分依赖于机器学习模型的自我学习能力,在对应的疾病数据上表现出色。然而,这些模型普遍存在缺乏泛化的能力,在其他疾病数据中表现不佳。
本研究采用宏基因组学和机器学习模型,利用多组独立的宏基因组数据进行研究。通过生物信息学工具对原始数据进行预处理,并使用数据降维和随机森林模型设定特征重要性阈值,筛选与疾病发生高度相关的特征菌群。接着,进行特征相关性分析,并使用加权平均的方法构建一种融合模型,旨在解决传统疾病筛查和诊断方法的侵入性和耗时问题,以及现有模型缺乏泛化能力的限制。本研究通过结合宏基因组学和机器学习技术,期望建立一种基于肠道菌群的非侵入性筛查和诊断模型,为疾病的早期筛查和诊断提供辅助参考,改善人们的健康状况。
1 材料与方法
1.1 数据获取及标准处理
从NCBI(美国国家生物技术信息中心)的SRA数据库中,选择3个样本的宏基因组测序数据,分别为Cirrhosis数据集(ERP005860)、T2D数据集(SRA045646、SRA050230、ERP002469)、Obesity数据集(ERP003612)。Cirrhosis数据集包括健康者118名、疾病患者114名;T2D数据集包括健康者217名、疾病患者223名;Obesity数据集包括健康者89名、疾病患者164名。
上述的3个样本数据集都采用人类微生物计划[13]所制定的标准,来预处理宏基因测序数据,首先使用FastQC和MultiQC对原始测序数据进行质量控制处理,接着利用工具KneadData对经过质量控制的序列去除宿主污染得到纯净序列,最后使用默认的参数在纯净的测序数据上运行MetaPhlAn2来生成物种组成表,测序数据处理流程如图1所示,宏基因组数据经过处理后得到的物种组成表如表1所示。
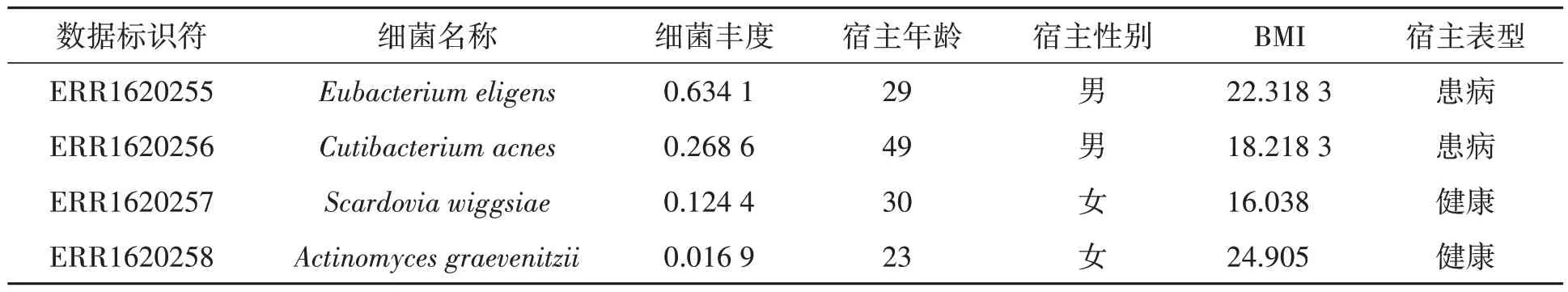
表1 部分原始数据Table 1 Part of raw data
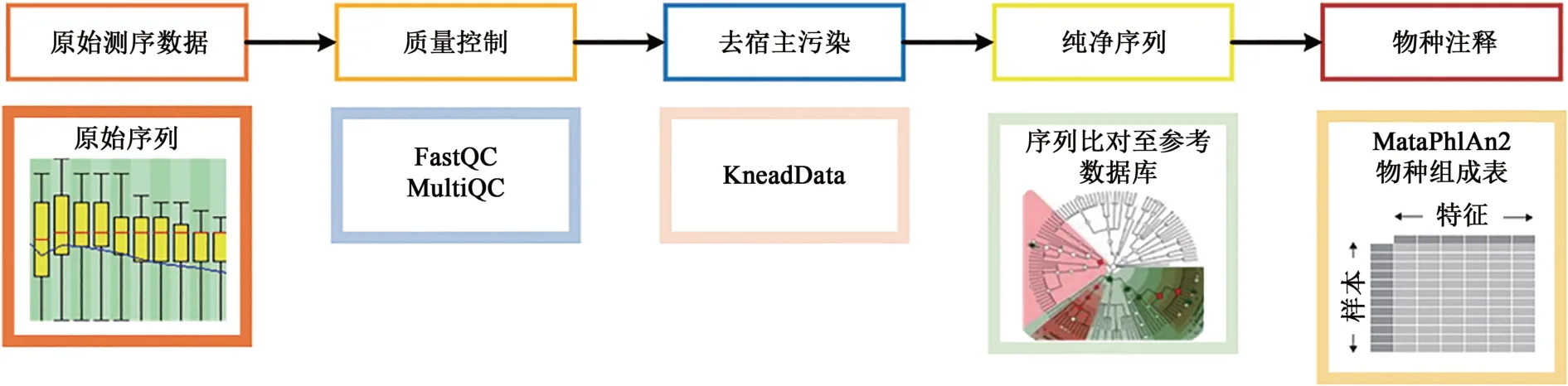
图1 测序数据处理流程图Fig. 1 Sequencing data processing flowchart
1.2 特征工程
由于宏基因测序数据包含成千上万个微生物DNA序列,并且每个微生物可能具有数千到数百万个基因。同时,每个样本的菌群组成也可能因样本来源、环境条件等因素存在差异,从而导致该类型的数据通常都是高维稀疏的,所以需要依据处理的数据集来对比选择合适的降维、筛选方法。为了对比数据降维和特征筛选对疾病预测模型精度的影响,本文使用主成分分析(principal component analysis,PCA)[14]、自编码器(AutoEncoder)[15]、非线性降维(T-SNE)[16]进行数据降维、对比,使用随机森林模型进行特征筛选,最后进行特征菌群的相关性分析。
1.3 加权平均融合模型的构建
加权平均融合模型(weighted average fusion model)是一种集成学习方法[17],通过对多个单模型的预测结果进行加权平均,从而得到最终的预测结果。通过对比子模型的预测结果和真实值之间的关系,设定权重值来降低模型融合后的预测误差。在分类问题中,加权平均集成算法在二分类和多分类问题上均表现出更高的预测精度。本实验使用支持向量机(support vector machine,SVM)[18]、极度梯度提升树(extreme gradient boosting,XGBoost)[19]、多层感知机(multilayer perceptron,MLP)[20]3个子模型进行预测,并采用加权平均来判定各子模型的预测结果与真实值的差异。本文使用了3种不同的加权融合方法,即软投票[21]、Stacking[22]和加权平均,并通过可视化方式进行预测结果的对比。比较T-SNE降维和随机森林特征选择后每种融合模型预测结果指标,分析了随机森林筛选出的特征菌群的重要程度,阐明各特征菌群在各个疾病发生发展过程中发挥的潜在性作用,实验的整体流程如图2所示。
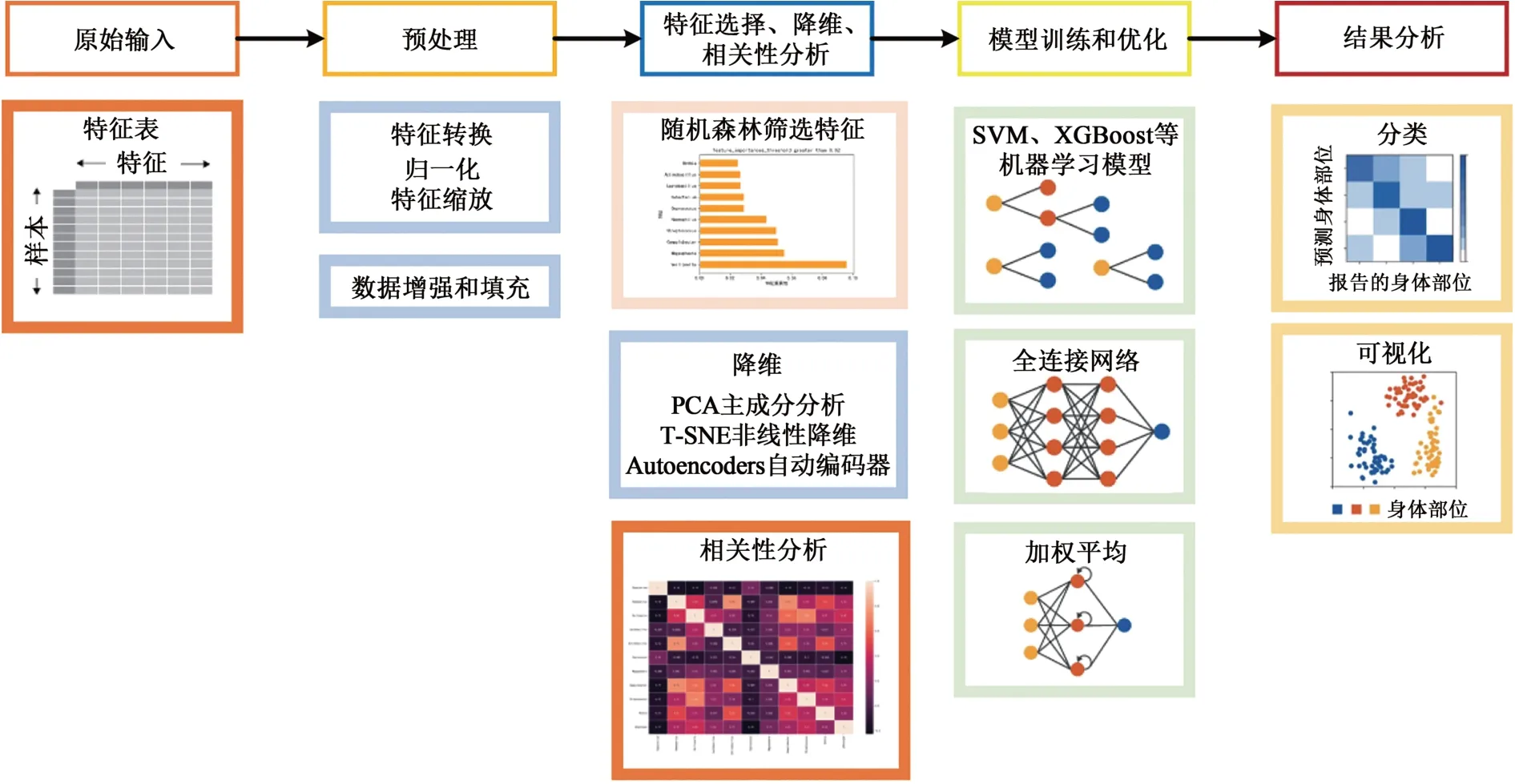
图2 实验流程图Fig. 2 Experimental flowchart
在加权平均融合模型中,每个单一模型的预测结果被赋予一个权重,权重的大小反映了这个模型的预测能力。一般来说,权重越大的模型对最终预测结果的贡献越大。
本文采用排序法是一种常见的加权平均模型融合技术,它可以基于单一模型在一些表现指标上的表现,为每个单一模型分配一个权重,以此来组合多个单一模型的预测结果,从而得到最终的预测结果。这种方法的基本原理是根据单一模型在各个表现指标上的表现来为它们赋予不同的权重,从而使得在预测结果中表现更好的单一模型能够产生更大的影响,而表现较差的单一模型则产生较小的影响。加权平均融合中基于表现指标的排序法的公式见式(1)。
其中,k是评价指标的数量,wj是第j个指标的权重,满足=1。
为了在加权平均融合中确定每个单一模型的权重,可以使用式(2)计算。
其中,n表示单一模型的数量,si表示第i个单一模型的总体得分,表示所有单一模型的总体得分之和。
具体来说,基于表现指标的排序法通常包含以下步骤:①选择一些表现指标,这些指标应该能够反映单一模型的预测能力,例如准确率、精度、召回率、F1分数等;②对每个单一模型在这些指标上进行评估,并计算它们在各个指标中的得分;③根据各个指标的重要性,为每个指标赋予一个权重;④对于每个单一模型,将它在每个指标上的得分乘以对应的权重,并对乘积求和,从而得到该单一模型的总体得分;⑤根据每个单一模型的总体得分,为它们分配一个权重,从而在加权平均融合中确定它们的贡献度。
本文选取的评价指标有精确率(precision)、准确率(accuracy)和ROC曲线下的面积(AUC),评价指标按公式(3)~(5)计算。
其中,TP表示真正例,TN表示真负例,FP表示假正例,FN表示假负例,rankinsi代表第i条样本的序号,M、N各自代表了正样本数量及负样本数量,∑insi∈positiveclass是遍历所有的正样本,并累加其序号。
2 结果与分析
2.1 特征降维和筛选
使用PCA、AutoEncoder、T-SNE 3种方法对3种疾病的物种组成表进行数据降维,接着可视化降维后3种疾病数据各个特征之间的分布(图3~5)。从降维后的特征分布图看出T-SNE的效果要好于PCA和AutoEncoder,经过AutoEncoder降维后的数据整体分布效果要好于PCA,这是因为T-SNE是一种非线性降维算法,相比于PCA,它可以更好地捕捉数据中的非线性结构。对于AutoEncoder来说,深度学习方法可以快速将高维数据降到低维,但由于AutoEncoder无法学习菌群特征之间的复杂结构,故在处理高维稀疏的宏基因测序数据时,通过T-SNE降维之后的数据分布更加均衡,最后将经过T-SNE降维后组成的新数据作为模型的输入数据。
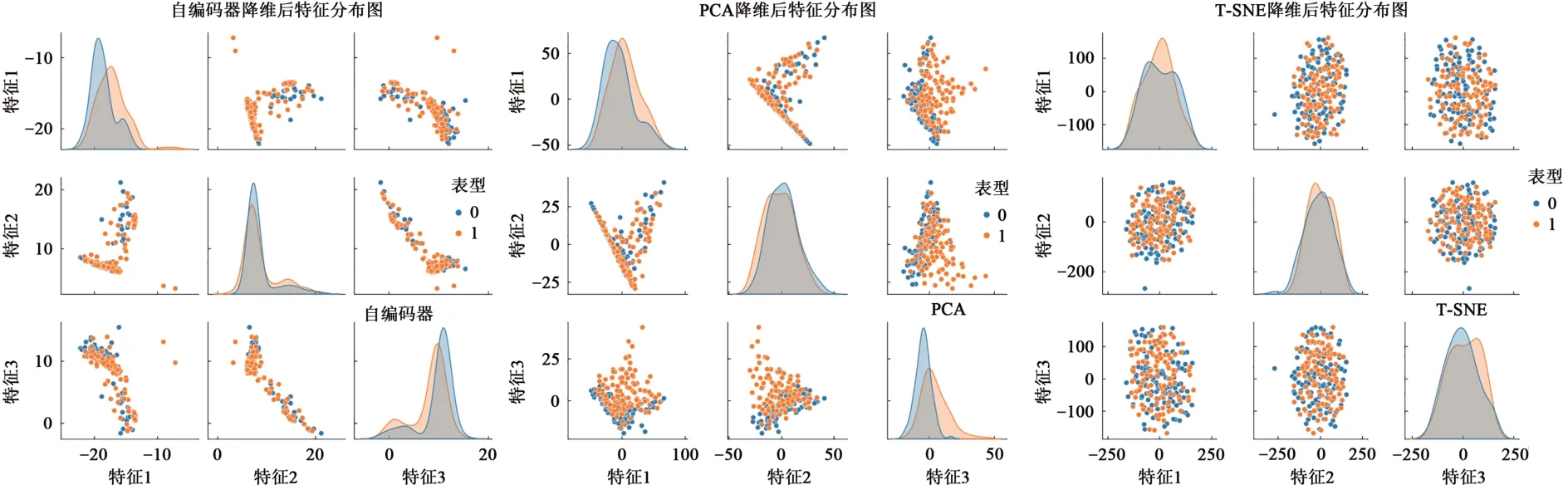
图3 肝硬化数据降维图Fig. 3 Dimensionality reduction diagram of liver cirrhosis data
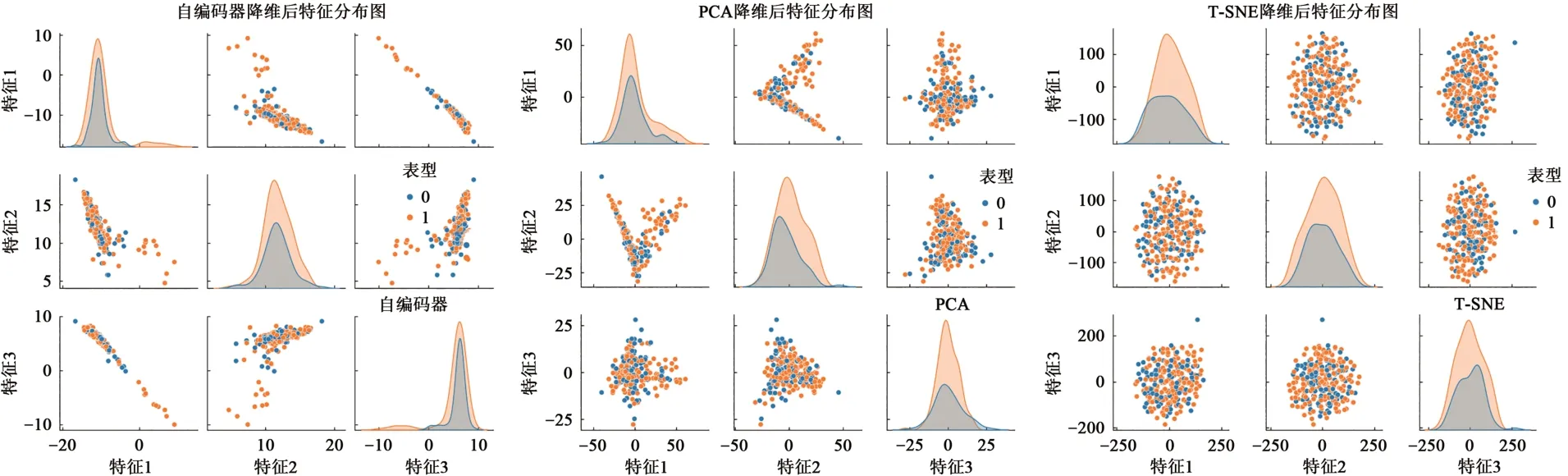
图4 肥胖症数据降维图Fig. 4 Dimensionality reduction diagram of obesity data

图5 糖尿病数据降维图Fig. 5 Dimensionality reduction diagram of diabetes data
使用随机森林模型对物种组成表进行特征筛选,设定好特征重要性阈值,将原始特征中重要性高于阈值的特征筛选出来,并由低到高呈现出来(表2),组成新的数据作为模型的输入,对比T-SNE降维产生的数据,观察是否对模型精度产生影响。
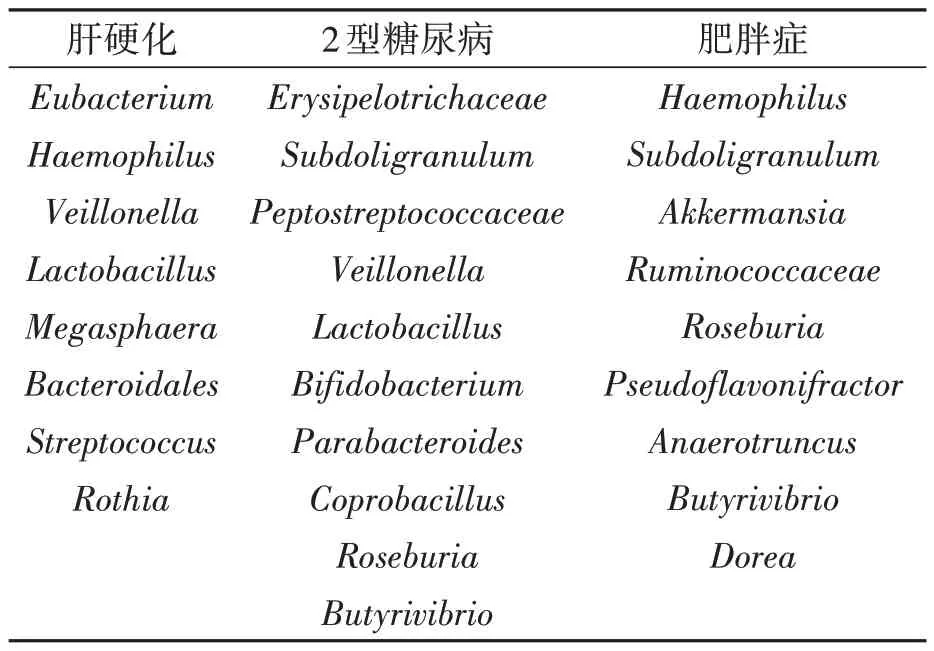
表2 3种数据经过特征选择后的特征菌群Table 2 Three types of data with characteristic microbial communities after feature selection
最后对降维后筛选出的数据进行相关性分析(图6),以便理解特征之间的关系,确定哪些特征菌群对目标变量的影响最大,这有助于建立更准确的预测模型。另外,检测多个特征菌群之间是否存在高度相关性[23],这种情况可能导致模型过拟合或不稳定,同时,需要注意避免选择过多的特征,以避免过拟合和降低模型的泛化能力。因此,在选择特征时,需要综合考虑各个特征菌群的相关性、重要性和可解释性等因素,选择最具有代表性的菌群进行建模。
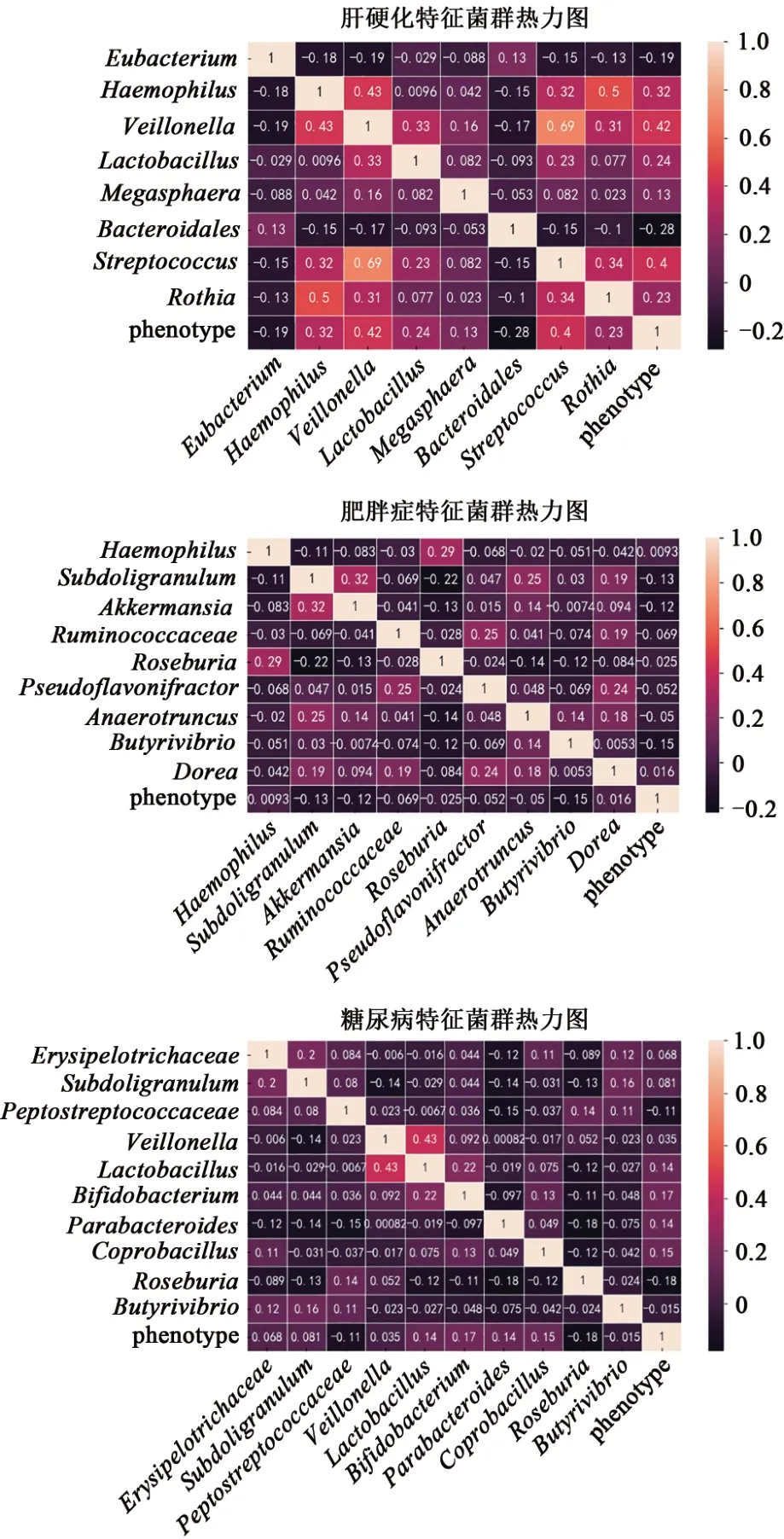
图6 3种疾病数据特征相关性分析Fig. 6 Correlation analysis of three disease data features
2.2 加权平均融合模型的评价
将加权融合模型与其他2种模型融合方法(软投票、Stacking)的性能进行了基准测试。使用肝硬化、2型糖尿病和肥胖症数据集交叉验证了模型的准确性,方便与现有的模型进行比较。在表3中,对数据使用T-SNE降维,加权平均模型在肝硬化数据集上执行的交叉验证的AUC值为0.5901,在2型糖尿病数据集上执行的交叉验证的AUC值为0.5651,在肥胖症数据集上执行的交叉验证的AUC值为0.3871。
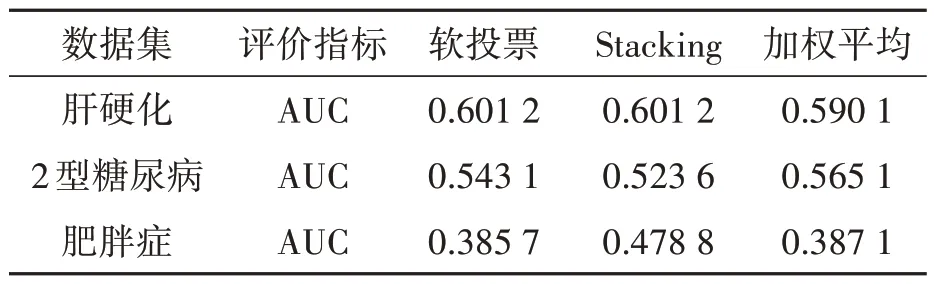
表3 经过T-SNE降维后模型在3种数据集上交叉验证的结果Table 3 Cross validation results of the model on three datasets after T-SNE dimensionality reduction
对于这些数据集,使用随机森林筛选数据特征,设定特征重要性阈值,将选择后特征作为模型的输入,在表4中,加权平均模型在肝硬化数据集上执行的交叉验证的AUC值为0.9286,在2型糖尿病数据集上执行的交叉验证的AUC值为0.6521,在肥胖症数据集上执行的交叉验证的AUC值为0.5747。
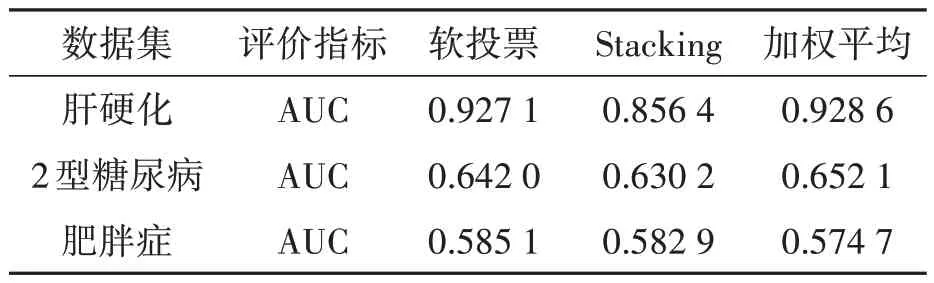
表4 经过特征选择后模型在3种数据集上交叉验证的结果Table 4 Cross validation results of the model on three datasets after feature selection
比较模型在经过T-SNE降维和随机森林特征筛选后的效果,发现经过降维后的数据特征会影响到分类模型原本的效果,可能是因为T-SNE对数据的处理比较复杂,需要调整的参数较多,容易出现“拥挤”问题,并且T-SNE只能处理连续型数据,不能处理离散型数据,而随机森林特征筛选则不受这个限制。因此,随机森林特征筛选相较于T-SNE降维具有更好的效果。
为了比较3种融合模型的拟合效果,对经过特征筛选和T-SNE降维的3种数据集中不同融合模型的AUC值进行数据可视化。从图7中可以看出,在使用随机森林进行特征筛选前加权平均的ROC曲线下的面积效果最好,经过随机森林模型的特征筛选后,软投票和加权平均的ROC曲线下的面积效果最好,故加权平均融合模型在进行特征选择前后的综合性能要优于其余融合模型,是3种融合方法中表现最佳的。
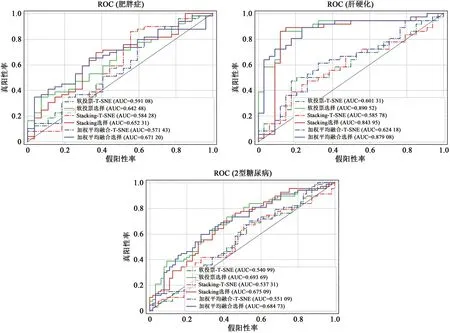
图7 融合模型ROC曲线下的面积图Fig. 7 Area chart under ROC curve of fusion model
本文使用多种模型进行宿主表型预测,包括3种单一子模型和3种融合模型,以及K近邻、逻辑回归2种适用于宿主表型预测的机器学习模型。单一模型的预测结果在表5中展示,这些对比分析旨在证明加权平均融合模型的准确性和有效性。
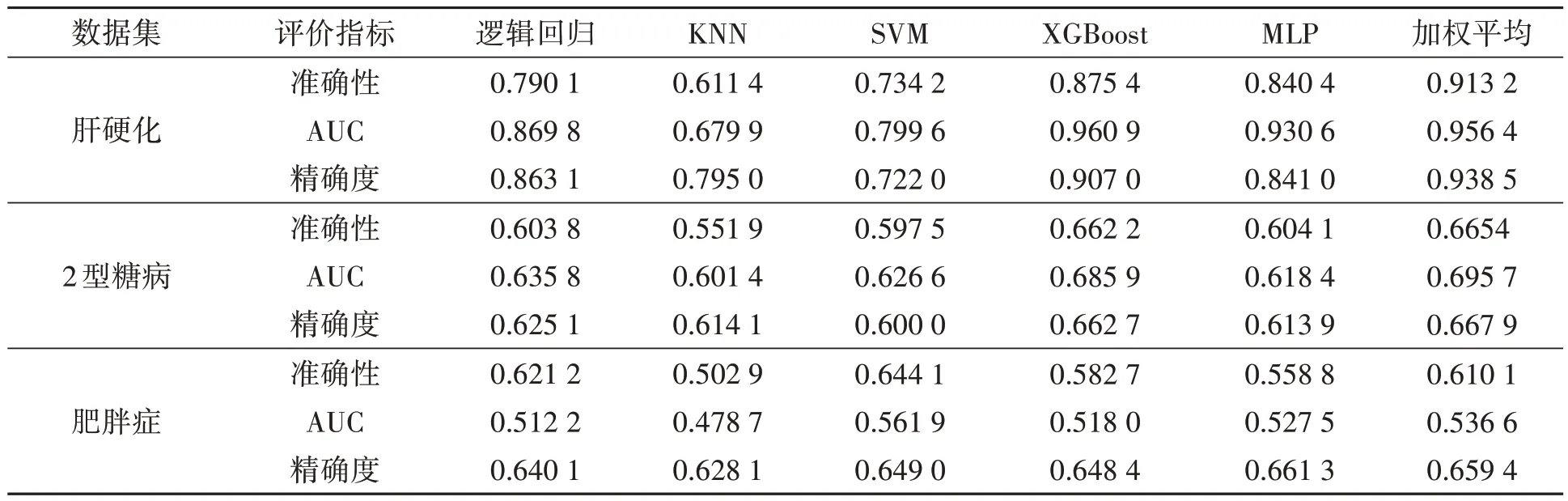
表5 单一模型预测结果Table 5 Prediction results of single model
2.3 加权平均模型中3种疾病数据各特征细菌的重要程度
使用随机森林算法来训练特征筛选模型,并根据特征的重要性绘制特征重要性图(图8),对于加权平均模型,每个特征的重要性可以通过对该特征的权重进行分析确定。在3种疾病数据中,每个特征代表不同的细菌。在确定每个特征的重要性时,要考虑以下3个因素。①权重大小。加权平均模型中每个特征的权重表示其在模型中的重要性。权重越大,特征对模型的影响越大。②相关性。特征之间的相关性也会影响其在模型中的重要性。如果2个特征高度相关,它们的权重可能会减少。③数据分布。特征在数据集中的分布也会影响其重要性。如果一个特征在数据集中出现的频率很高,那么它对模型的影响可能会更大。
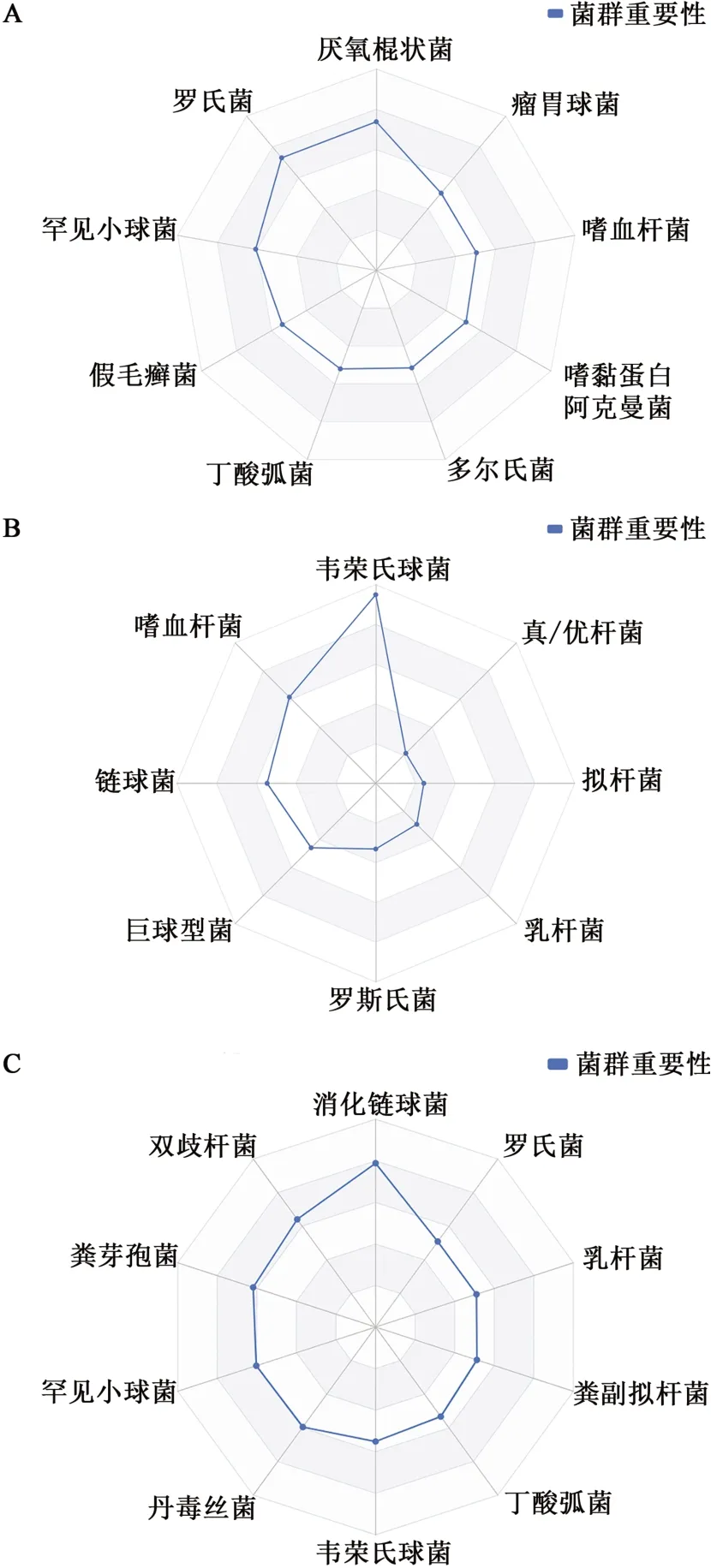
图8 3种数据在模型中特征细菌的重要性Fig. 8 The importance of three types of data in characteristic bacteria in models
从图8可以看出,在肝硬化数据中韦荣氏球菌、嗜血杆菌、链球菌等特征菌群的重要程度较高,消化链球菌、双歧杆菌、粪芽孢菌等特征菌群在糖尿病数据中占有较高的特征重要性,而对于肥胖症数据,厌氧棍状菌、罗氏菌、罕见小球菌等特征菌群具有较高的重要性。此外,筛选出的特征菌群并不只存在于一种疾病中,特定的菌群可以与多种疾病相关联,因此在研究微生物菌群与疾病之间的关系时,要考虑不同疾病之间微生物群落的共性和差异性,从而可以更好地理解微生物群落与疾病之间的关系。
3 讨论
在3种疾病数据中,使用随机森林的方法能够筛选出多种特征菌群,比如消化链球菌、厌氧棍状菌和韦荣氏球菌。较之于2型糖尿病患者,消化链球菌可能更有益。2型糖尿病是一种慢性疾病,通常与胰岛素抵抗和胰岛素缺乏有关。胰岛素抵抗意味着身体无法有效地利用胰岛素,而胰岛素缺乏则可能导致血糖水平升高。研究表明,消化链球菌可以代谢产生乳酸,而这种酸可以提高胰岛素敏感性[24]。对于肥胖症来说,厌氧棍状菌是一种有益的肠道菌群,它们在缺乏氧气的环境中生长繁殖。研究表明,肥胖症患者肠道中的厌氧棍状菌数量减少,这可能导致代谢疾病的产生。而通过增加厌氧棍状菌的摄入量,可以改善肥胖症患者的肠道菌群平衡,降低代谢疾病的风险[25]。另外,韦荣氏球菌可能有助于改善肝硬化患者的健康状况。一项研究表明,肝硬化患者肠道中的韦荣氏球菌数量减少,这可能导致肠道屏障的受损和炎症反应的增加[26]。需要注意的是,目前关于特征菌群在对应疾病上发挥的作用还需要更多的研究来证实特征菌群对疾病患者的实际益处。这些研究都证明了本文基于随机森林方法筛选出多种特征菌群的基础上,构建融合疾病诊断模型的合理性。
本研究通过对3种疾病的宏基因测序数据进行疾病预测建模和特征筛选,分别选择出与各种疾病发生发展相关的菌群特征。基于随机森林模型筛选出的特征菌群,再使用加权平均融合方法,建立不同疾病的辅助诊断模型,同时对比了MetAML工具[9]所使用的SVM分类模型,尽管在肥胖症数据集上,该模型的AUC值要大于加权平均模型。但在肝硬化和糖尿病数据集上,该模型的AUC值要小于加权平均模型。对比发现SVM模型在不同的数据集上的模型预测能力要稍弱于加权平均模型。同时,加权平均融合模型选择的特征菌群的预测准确率相较于投票法和Stacking融合方法是最高的,在3种疾病数据划分的训练集和测试集上均能保持相对较好的预测准确率。使用加权平均融合模型,可以做到进一步联系肠道菌群与患者表型,再结合随机森林方法构建的特征菌群筛选模型,进一步加快特征菌群在日常疾病筛查诊断上的使用。总体来说,加权平均模型为识别与疾病相关的微生物特征和开发用于早期检测和预防相关疾病的非侵入性诊断工具提供了一种辅助方法。
本文在研究过程中仍存在一定的不足,例如所获取的疾病数据量有限,未能充分考虑疾病数据样本量对模型的影响。因此,下一步的研究需要扩大数据收集的疾病种类,以涵盖更广泛的疾病,从而提高融合模型的覆盖面和适用性,更全面地验证融合模型的效果,提高模型的准确性和泛化能力。
猜你喜欢
车主之友(2022年4期)2022-08-27
中老年保健(2022年2期)2022-08-24
科学(2020年4期)2020-11-26
数学年刊A辑(中文版)(2020年2期)2020-07-25
海峡姐妹(2019年12期)2020-01-14
数学物理学报(2019年6期)2020-01-13
数学物理学报(2017年5期)2017-11-23
动物营养学报(2015年10期)2015-12-01
现代检验医学杂志(2015年4期)2015-02-06
计算物理(2014年1期)2014-03-11
