
基于生物信息学识别早期肺腺癌生物标志物*
2023-10-11 01:09苗桂华袁金珊李德涛侯玉磊
国际检验医学杂志 2023年19期
苗桂华,袁金珊,李德涛,侯玉磊,陈 辉
重庆医科大学附属第一医院医学检验科,重庆 400016
肺癌是常见的恶性肿瘤,是导致癌症死亡的主要原因[1]。肺腺癌约占所有肺癌的40%。由于吸烟的减少和药物治疗的改进,肺腺癌的病死率有所下降,但5年生存率仍不容乐观[1-2]。低剂量CT(LDCT)降低了早期肺腺癌患者的死亡风险,但因为扫描辐射及较高的假阳性,专家建议不将LDCT作为肺腺癌普查项目,仅用于高危人群筛查[3]。肺腺癌风险预测模型是识别早期肺腺癌的有效手段之一,在此基础之上针对高危人群进行的LDCT,可有效避免过度诊断和假阳性结果。因此,在肺腺癌筛查实际应用中,亟需补充符合早期筛查要求的风险预测模型来选择高危人群,必要时再行LDCT筛查。近期研究表明,可通过生物标志物建立有效的风险预测模型,在LDCT之前通过实验室检测进行肺腺癌风险评估[4]。
由于人类基因多样性、环境差异及种族不同的影响,单个实验难以得到与肺腺癌相关的确切基因。目前,通过测序技术可以对肿瘤组织进行核酸测序,然后比较肿瘤组织和正常组织之间的差异表达基因(DEGs)。基于微阵列和测序的生物信息学技术已广泛应用于DEGs筛选,这也为包括肺腺癌在内的恶性肿瘤生物标志物的获取提供了便利。通过生物信息学筛选肺癌中的DEGs,可以扩大样本量并提高结果的可信度。然而,目前已发表的研究仍有一些缺陷,如在选择样本的标准上并不严格。HE等[5]使用基因表达数据库(GEO)数据集GSE43458和肿瘤基因组图谱(TCGA)数据来识别肿瘤组织和正常组织之间的DEGs,确定VWF为肺癌的关键基因和潜在的生物标志物,但其所选的数据集包含Ⅲ期和Ⅳ期肺癌数据,尚不能满足早期诊断的要求。本研究拟选取的GEO数据集将限定在早期肺腺癌,通过GEO2R在线分析工具获得共同表达的DEGs,使用DAVID在线分析工具进行基因本体论(GO)和京都基因与基因组百科全书(KEGG)富集分析,使用STRING和Cytoscape构建和可视化蛋白质-蛋白质相互作用(PPI)网络,采用cytoHubba选择PPI网络的中枢基因,并使用GEPIA2数据库分析中枢基因的mRNA表达水平和总生存率(OS),以期从具有代表性的数据集中寻找肺腺癌早期诊断的生物标志物。
1 资料与方法
1.1数据的获取 在GEO数据库[6]中查询“肺腺癌(LUAD)”和“非小细胞肺癌(NSCLC)”。在所得结果中进一步确定选择数据集的标准如下:(1)应含有肺腺癌组织和相应的邻近正常组织;(2)所有选定的肺腺癌病例均应诊断为Ⅰ期或Ⅱ期肺腺癌;(3)数据集的最近更新日期在5年以内。因此,从GEO数据库中选择了GSE27262、GSE63459、GSE116959和GSE118370。GSE27262(平台GPL570)包含25例肺腺癌Ⅰ期患者的配对的肿瘤和邻近正常组织。GSE63459(平台GPL6883)包含33个肺腺癌Ⅰ期患者的肿瘤组织和32个正常组织。GSE116959(平台GPL17077)包含57个Ⅰ期或Ⅱ期肺腺癌组织和11个相应的邻近正常组织。GSE118370(平台GPL570)包含6对肺腺癌Ⅰ期或Ⅱ期患者的肿瘤组织和正常组织。所选数据集包含来自美国、中国、法国的样本,相比其他研究更具代表性。这些数据集的临床特征见表1。

表1 所选择数据集的临床特征
1.2识别差异表达的基因 采用GEO2R计算各个数据集肺腺癌肿瘤组织和正常组织样本之间的DEGs。GEO2R是基于R语言的limma算法比较两组样本的不同。首先,设置GEO2R,根据调整后的P<0.05和倍数变化(FC)的对数的绝对值|log2FC|>1的标准筛选出DEGs。然后,绘制这些数据集DEGs的Venn图。最后,选择所有数据集共同表达的DEGs来进一步研究。
1.3基因注释和途径分析 DAVID在线分析工具可对共同DEGs进行GO和KEGG注释[7]。GO分析从生物过程、细胞成分和分子功能3个方面对基因进行解释。KEGG则分析基因可能参与的代谢途径。以P<0.05为差异有统计学意义。
1.4PPI网络的建设 STRING数据库和Cytoscape软件用于构建和可视化共同DEGs的PPI网络[8]。随后通过使用cytoHubba程序中的最大集团中心度(MCC)算法,选取该算法得分最高的10个基因作为中枢基因,得到PPI关系最紧密的中枢基因。
1.5中枢基因的表达及生存分析 应用GEPIA2中的基因表达数据库验证中枢基因的实际表达情况,并探索中枢基因与患者OS的关系。采用Mantel-Cox检验选择对肺腺癌患者OS有显著影响的基因作为潜在的肿瘤标志物。
2 结 果
2.1共同表达的DEGs 由GSE27262数据集确认的DEGs包括925个上调和1 374个下调的DEGs,由GSE63459数据集确认的DEGs包括98个上调和309个下调的DEGs,由GSE116959数据集确认的DEGs包括752个上调和1 506个下调的DEGs,由GSE118370数据集确认的DEGs包括245个上调和688个下调的DEGs。各个GEO数据集差异表达基因的火山图见图1A。通过Venn图的结果发现,这些数据集有154个共同的DEGs,包括15个上调和139个下调的DEGs,见图1B。所有数据集共同的DEGs具体如下。(1)表达上调的基因:GOLM1、PROM2、HS6ST2、TOP2A、C1orf106、UBE2T、SPDEF、HMGB3、NME1、GJB2、TIMP1、SULF1、FAM83A、SPINK1;(2)表达下调的基因:AGER、MME、SPTBN1、SPOCK2、SH3GL3、PTPRB、BTNL9、GRK5、EDNRB、CDO1、FAM107A、LIMS2、TEK、FAM189A2、STXBP6、STARD13、SH2D3C、SLC6A4、DACH1、FABP4、STX11、TNNC1、CAV1、EPAS1、NTNG1、RASIP1、FGD5、HIGD1B、CRYAB、LIMCH1、S1PR1、GIMAP8、ACVRL1、CAV2、COX4I2、TGFBR3、FOXF1、LDB2、FCN3、TBX3、NOTCH4、VGLL3、CD36、ADARB1、SASH1、SDPR、SLIT2、TCF21、CLEC14A、FERMT2、CD93、ARAP3、FHL1、TACC1、FMO2、ANKRD29、TMEM47、SVEP1、PECAM1、PLAC9、PTRF、TMEM204、CD300LG、ACADL、JAM2、SOSTDC1、COX7A1、CLIC5、ADH1B、PPP1R14A、FZD4、CLEC1A、GHR、LMCD1、FXYD6、VWF、AHNAK、FEZ1、CYYR1、LHFP、KLF4、CALCRL、PDE5A、ESAM、PLEKHH2、MYH10、KLF9、ABI3BP、VIPR1、GNG11、ANGPT1、NDRG2、WISP2、SMAD6、NDRG4、RAMP3、PMP22、TMEM100、CLDN18、ADRB2、WFDC1、PDK4、TBX2、COL13A1、MFAP4、GBP4、PODXL、BMPR2、HBB、FGR、ENG、SPARCL1、MACF1、SFTPC、HYAL1、CAMK2N1、GPC3、HBEGF、TIMP3、LRRC32、DKK3、MAL、HOXA5、ALOX5、IL1RL1、C1orf162、PDZD2、APOL3、FBLN1、DUOX1、AQP4、FPR1、CD52、GPX3、MYH11、SLCO2A1、SLC1A1、TCEAL2、CA2。

注:A为GSE27262、GSE63459、GSE116959和GSE118370的DEGs火山图;B为4个数据集中表达上调、表达下调及全部的共同DEGs。图1 从GEO数据库中识别肺腺癌中共同的DEGs
2.2GO和KEGG分析 通过DAVID对所有上调和下调的共同DEGs进行富集分析,结果显示,所有的共同DEGs在生物过程中的血管新生、分支结构的形态形成和白细胞迁移等过程中显著富集;共同DEGs的细胞成分聚集区域是膜筏、质膜的组成成分和细胞外空间的位置等;共同DEGs的分子功能富集于转化生长因子-β(TGF-β)结合、TGF-β激活受体活性和肝素结合等,见图2A。KEGG富集分析显示,共同DEGs参与癌症中的蛋白聚糖、细胞黏附分子和神经活性配体-受体相互作用等代谢途径中,见图2B。

注:A为GO分析后P<0.05的前10位结果;B为KEGG富集分析后取P<0.05的前5位结果。图2 GO和KEGG分析结果、PPI网络构建结果及获取的中枢基因
2.3PPI网络中枢基因 通过STRING数据库和Cytoscape软件绘制所有的共同DEGs的PPI网络,由154个节点和195条边组成。利用cytoHubba程序的MCC算法,选择MCC得分最高的前10位基因(PECAM1、ENG、VWF、TEK、TIMP1、CAV1、ANGPT1、ACVRL1、BMPR2和SMAD6)作为中枢基因,中枢基因相互作用关系图,见图3。MCC得分越高代表该蛋白质与其他蛋白质的PPI关系越紧密,见表2。
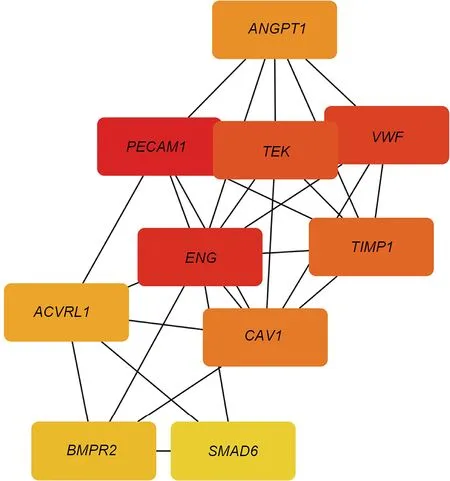
注:红色到黄色的节点代表中枢基因的连接关系由强到弱。图3 共同DEGs的中枢基因及相互作用关系

表2 MCC算法得分前10位的中枢基因
2.4中枢基因的验证及生存分析 GEPIA2在线分析工具的mRNA表达水平验证结果显示,除TIMP1外,其余9个中枢基因在肺腺癌患者中均表达下调,见图4A,验证的结果与前文的研究结果一致。此外,通过GEPIA2进行了中枢基因在肺腺癌患者的OS分析见图4B。根据所得到的生存分析结果进行Mantel-Cox检验发现,PECAM1和ENG的低表达与预后不良相关(P<0.05),见图4C。最终,PECAM1和ENG被选为诊断和预后的潜在生物标志物。

注:A为使用GEPIA2的数据库验证肺腺癌肿瘤组织和正常组织中PECAM1、ENG、VWF、TEK、TIMP1、CAV1、ANGPT1、ACVRL1、BMPR2和SMAD6的mRNA表达水平;B为 PECAM1、ENG、VWF、TEK、TIMP1、CAV1、ANGPT1、ACVRL1、BMPR2和SMAD6 的mRNA表达水平对肺腺癌患者的预后影响;C为使用Mantel-Cox检验比较肺腺癌中不同基因对生存预后影响的贡献度,由蓝到红表示影响程度由强到弱。图4 中枢基因的验证及生存分析结果
3 讨 论
肺腺癌是最常见的恶性肿瘤之一,中晚期肺癌患者往往预后不良,缺乏早期诊断肺腺癌的生物标志物是重要原因之一。本研究选择早期肺腺癌的GEO数据集,通过生物信息学方法来确认与早期肺腺癌患者相关的中枢基因,期望得到可用于早期肺腺癌患者筛选和改善预后的生物标志物。
本研究中筛选的4个GEO数据集GSE27262、GSE63459、GSE116959和GSE118370,包含来自不同地区的数百个Ⅰ期或Ⅱ期样本,既考虑了方法、环境和种族等差异的影响,又将研究对象限定在早期肺腺癌中。通过比较发现,4个数据集的DEGs为15种上调基因和139种下调基因。DAVID在线分析工具分析结果显示,这些DEGs主要富集于血管新生、细胞膜筏、TGF-β结合及癌症的蛋白聚糖相关基因。同时,使用cytoHubba程序获得10个中枢基因,分别是PECAM1、ENG、VWF、TEK、TIMP1、CAV1、ANGPT1、ACVRL1、BMPR2和SMAD6,其中仅TIMP1在早期肺腺癌中表达上调,其余9个基因均为表达下调。生存分析结果显示,PECAM1和ENG的表达水平降低与肺腺癌患者生存预后不良相关,值得进一步研究。GEPIA2是一款广泛用于分析基因表达和分析肿瘤组织与正常组织样本之间的相互作用的在线分析工具[9],结果经验证后可极大地提高可信度。本研究中,GEPIA2在线分析工具的mRNA表达水平验证结果显示,除TIMP1外,其余9个中枢基因在肺腺癌患者中均表达下调,验证结果与GEO2R的筛选结果一致,根据所得到的生存分析结果进行Mantel-Cox检验发现,PECAM1和ENG低表达与肺腺癌患者预后不良相关(P<0.05)。
PECAM1,也称为CD31,主要在血小板、单核细胞、中性粒细胞和特定T细胞的表面发现[10],参与内皮细胞的细胞间连接[11]。PECAM1在血管生物学中的基本作用是可参与嗜异性或同质性结合。有研究表明,PECAM1与整合素激活、血管生成和白细胞迁移有关[12]。此外,PECAM1也是组织中早期血管成分的重要标志物。NEWMAN等[13]发现,PECAM1可与TGF-β受体(TGF-βR)发生物理相互作用,并干扰体内CD8+T细胞介导的肿瘤清除过程。CHEN等[14]发现,在转移了A549细胞的严重联合免疫缺陷小鼠中,PECAM1可促进肺癌的进展和转移。TAKASE等[15]报道,PECAM1在正常组织的染色结果高于肿瘤组织,而近期的一项研究发现,PECAM1也在其他的癌症中存在表达差异,如早期喉鳞状细胞癌和血管肉瘤[16-17]。这些研究结果与本研究基于生物信息学的结果一致。
ENG是血管内皮的一种糖蛋白,可与TGF-βR的β1和β3肽结合[18]。ENG作为一种多效性细胞因子,具有调节细胞迁移、增殖和分化等不同的功能[19]。肿瘤生长与血管形成高度相关,因此组成血管内皮的ENG往往是检测肿瘤特异性新生血管的重要标志物[20]。 KUMAR等[21]通过免疫组化分析发现,血管增殖和形成在肿瘤生长和侵袭中具有重要作用。近年来,胆管癌和胶质母细胞瘤等多种癌症已被发现与ENG的表达水平有关[22-23]。此外,在癌症患者的血液中也检测到可溶性ENG[24]。这些结果与生物信息学分析得出的ENG与早期肺腺癌,尤其是与血管生成相关是一致的。上述研究结果共同表明,ENG可作为早期肺腺癌的有效生物标志物。
有研究报道,PECAM1/ENG比值是评价血管生成的重要指标。因为PECAM1可用于识别内皮细胞,而ENG可识别活化的内皮细胞。 BAUMAN等[25]报道显示,在肾细胞癌中,PECAM1/ENG比值与肿瘤分期一致(P<0.000 1)。 MARIONI等[26]报道显示,PECAM1/ENG可用于评估喉鳞状细胞癌的预后。因此,有望将PECAM1/ENG比值用于评估肺腺癌的肿瘤进展。
综上所述,本研究选取来自不同地区的早期肺腺癌患者的4个GEO数据集,通过生物信息学分析发现,DEGs主要与血管新生、细胞膜筏、TGF-β结合和癌症蛋白聚糖相关,生存分析表明,PECAM1和ENG的表达下调与肺腺癌患者预后不良具有相关性。本研究结果表明,PECAM1和ENG在早期肺腺癌的诊断和进展中具有重要作用,可作为肺腺癌诊断和预后判断的潜在生物标志物。
猜你喜欢
辽宁省博物馆馆刊(2020年0期)2020-08-13
中成药(2018年7期)2018-08-04
中华老年多器官疾病杂志(2016年9期)2016-04-28
西南军医(2016年3期)2016-01-23
淮海医药(2015年1期)2016-01-12
医学研究杂志(2015年7期)2015-06-22
中国当代医药(2015年22期)2015-03-01
中国当代医药(2015年17期)2015-03-01
现代检验医学杂志(2015年6期)2015-02-06
现代检验医学杂志(2015年5期)2015-02-06
