
医院制剂上感合剂微生物限度检查方法建立
2023-10-10 03:44赵婷曾博雅马佩杰卢俊玮舒文将强羽菲鞠依珊
贵州医药 2023年10期
赵婷 曾博雅 马佩杰 卢俊玮 舒文将 强羽菲 鞠依珊△
(1.宝鸡市中心医院,陕西 宝鸡 721008;2.宝鸡市食品药品检验检测中心,陕西 宝鸡 721013)
医院制剂主要是针对本院临床上需求量小、不适合进行工厂批量化生产的品种,控制其质量是确保临床用药安全的前提[1]。可以对药品质量产生影响的因素有很多,作为重要影响因素之一的微生物,对其进行控制是保证药品质量的重要手段[2]。控制菌检查法及微生物计数检查法是检查非规定灭菌制剂与其原辅料受微生物污染程度的重要方法[3]。口服中药制剂作为目前常用的医院内医疗制剂,具有用量大、制作简单及使用便捷等特点,且因其疗效确切深受患者与医生好评,由于中药制剂的特殊性,致使其无法像西药具有严格标准的制作流程,因此对医院内中药制剂的微生物限度进行检查常常不合格[4]。由于中药制剂中含有多种成分,其中任何成分均可对微生物限度检查结果的准确性产生影响,因此药品微生物限度检查所使用的方法应该与其他分析方法一样被证明是适宜的,才可以确保检查方法的准确完整[5-6]。微生物方法学试验即向药物制剂中加入一定量的试验菌,验证检验方法是否实用和有效[7]。上感合剂为我院院内特色的口服中药制剂,因其疗效确切、临床使用广泛,为对其质量进行控制,确保检测方法的可靠性,使其微生物限度达到要求[8]。本研究根据2015年版《中国药典》四部通则规定,对我院院内制剂上感合剂进行微生物限度检查的方法学验证试验,建立了该制剂微生物限度的检查方法。
1 材料与方法
1.1主要仪器 BHC-1300ⅡA2型生物安全柜(苏净集团苏州安泰空气技术有限公司);SPX-150型生化培养箱(慧科电子有限公司);KB-240型生化培养箱(德国BINDER公司);YXQ-LS-100SⅡ型立式压力灭菌器(上海博讯实业有限公司医疗设备厂);BSA2202S电子天平(赛多利斯科学仪器有限公司);VITEK2全自动微生物鉴定药敏系统法国梅里埃有限公司);Olympus显微镜(日本奥林巴斯公司)[9]。
1.2材料
1.2.1供试品 上感合剂(批号:190527,规格:200 ml/瓶),由我院制剂室提供。
1.2.2菌种 大肠埃希菌[CMCC(F)44102]、铜绿假单胞菌[CMCC(B)10104]、金黄色葡萄球菌[CMCC(B)26003]、枯草芽孢杆菌[CMCC(B)63501][10-15]、 黑曲霉[CMCC(F)98003]、白色念珠菌[CMCC(F)98001][1,6]。以上试验用菌株均为第3代,且来自于中国医学细菌保藏管理中心。
1.2.3培养基及稀释液 胰酪大豆胨液体培养基(批号:1703082)、沙氏葡萄糖液体培养基(批号:180420)、麦康凯液体培养基(批号:180711)、胰酪大豆胨琼脂培养基(批号:1703232)[1,8-9,14]、沙氏葡萄糖琼脂培养基(批号:180301)、麦康凯琼脂培养基(批号:180207)、PH7.0氯化钠-蛋白胨缓冲液(批号:1806222)。以上培养基均由北京三药科技开发公司生产,适用性检查符合要求。
1.3方法
1.3.1菌液的制备 分别将大肠埃希菌、铜绿假单胞菌、枯草芽孢杆菌及金黄色葡萄菌接种于胰酪大豆胨液体培养基中,置于33℃培养箱中,培养20 h[5-6,10-11,14,16-19],用PH7.0氯化钠-蛋白胨缓冲液制成含菌量为103~104cfu/mL的混悬液。将白色念珠菌接种于沙氏葡萄糖液体培养基中,置于23℃培养箱中,培养3 d[10,14,16],用PH7.0氯化钠-蛋白胨缓冲液制成含菌量为103~104cfu/mL的混悬液。将黑曲霉接种于沙氏葡萄糖琼脂斜面培养基中,置于23℃培养箱中,培养5 d,加入5 mL含0.05%(ml/ml)聚山梨酯80的PH7.0氯化钠-蛋白胨缓冲液,洗脱孢子,收集孢子悬液,制成含霉菌孢子数为103~104cfu的黑曲霉孢子混悬液。
1.3.2培养基稀释法制备供试液 取两瓶上感合剂,混合均匀,作为供试品原液。量取10 mL供试品原液,加100 mL胰酪大豆胨液体培养基,振摇,使其分散均匀,制成1:10的供试液。
1.3.3需氧菌、霉菌和酵母菌总数计数方法学验证试验 (1)试验组:量取制备好的1:10供试液分装于5个无菌试管中,每管分别为9.9 mL,再分别加入制备好的含菌量为103~104cfu/mL的枯草芽孢杆菌、铜绿假单胞菌、白色念珠菌、金黄色葡萄球菌及黑曲霉菌悬液0.1 mL,含菌量不大于100 cfu,混匀[10,12,16]。(2)供试品对照组:量取制备好的1:10供试液9.9 mL,加入0.1 mL PH7.0氯化钠-蛋白胨缓冲液,混匀。(3)菌液对照组:量取PH7.0氯化钠-蛋白胨缓冲液分装于5个无菌试管中,每管分别为9.9 mL,操作同试验组。(4)阴性对照组:用PH7.0氯化钠-蛋白胨缓冲液代替菌液及供试品,操作同试验组。(5)平皿倾注法对供试品微生物回收:量取1 mL制备好的试验组供试液,置于直径90 mm的无菌平皿中,注入20 mL温度不超过45℃熔化的胰酪大豆胨琼脂培养基或沙氏葡萄糖琼脂培养基,混匀,待凝固,胰酪大豆胨琼脂培养基倒置于33℃培养箱中培养5 d,沙氏葡萄糖琼脂培养基倒置23℃培养箱中培养7 d,分别进行计数。供试品5种试验菌的回收率均需进行3次重复试验,计算平均菌落数[9-10]。同法对菌液对照组、供试品对照组及阴性对照组的菌落数进行测定。
1.3.4控制菌大肠埃希菌的检查方法学验证试验 (1)试验组:量取1:10供试液10 mL接种于胰酪大豆胨液体培养基100 mL中,同时接入大肠埃希菌含菌量不大于100 cfu,混匀,置于33℃培养箱中培养18 h[10]。(2)供试品对照组:量取10 mL 1:10供试液,以稀释液PH7.0氯化钠-蛋白胨缓冲液代替菌液,操作同试验组。(3)阴性对照组:以稀释液PH7.0氯化钠-蛋白胨缓冲液代替菌液与供试液,操作同试验组[8]。(4)选择和分离培养:分别量取1 mL上述三组培养物接种至100 mL麦康凯液体培养基,置于43℃培养箱中培养24 h,将麦康凯液体培养物划线接种于麦康凯琼脂培养基平皿上,置于33℃培养箱中培养18 h,观察[10,14,16-17]。
2 结 果
2.1需氧菌、霉菌和酵母菌总数计数方法学验证试验结果 结果判定:回收率(%)=(试验组平均菌落数-供试品对照组平均菌落数)/菌液对照组平均菌落数×100%[20-21],若回收率在0.5~2.0范围内[22],上感合剂的需氧菌、霉菌和酵母菌总数计数的方法学验证试验合格[11,14,16,19]。结果显示:本批次上感合剂,采用培养基稀释法进行测定铜绿假单胞菌、白色念珠菌、金黄色葡萄球菌、黑曲霉、枯草芽孢杆菌的回收率均在0.5~2.0范围内[22],提示上感合剂的需氧菌、霉菌和酵母菌总数计数检查方法学验证合格[8-10,12,15-16]。见表1、表2。
2.2控制菌大肠埃希菌的检查方法学验证试验结果 结果判定:若菌液组能检出大肠埃希菌,供试品对照组与阴性对照组均未检出,可判定上感合剂的控制菌大肠埃希菌的检查方法验证试验合格[15]。结果显示:供试品对照组与阴性对照组均未检出试验菌,而试验组检出试验菌,检出的试验菌经鉴定为大肠埃希菌,提示上感合剂的控制菌检查方法验证合格。见表3、表4。
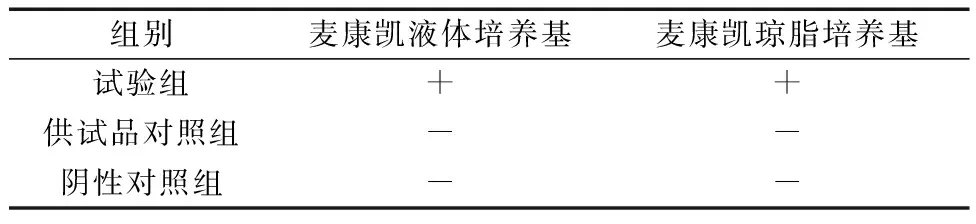
表3 控制菌(大肠埃希菌)检查方法学验证试验结果Ⅰ

表4 控制菌(大肠埃希菌)检查方法学验证试验结果Ⅱ
3 讨 论
微生物限度检验需确认供试品在该检验条件、检验量下无抑菌活性或者抑菌活性已经被充分消除,才可以保证检验结果能真实的反映制剂受微生物污染的情况[23]。但由于中药制剂通常成方药味较多,药不同抑菌活性强度不同,检验具有较强抑菌活性的制剂时,试验过程中首先选择方便有效的培养基稀释法,医院制剂上感合剂中含有连翘、金银花等中药,且金银花含有的绿原酸成分具有极强的抗菌及抗病毒功能[24-26],连翘作为广谱抗菌药,可抑制多种细菌[27],但表1、2试验结果显示,含有连翘、金银花的上感合剂对药典规定验证用的5种试验菌株(铜绿假单胞菌、金黄色葡萄球菌、白色念珠菌、枯草芽孢杆菌、黑曲霉)抑菌作用较小,且回收率均在0.5~2范围内[22],这可能是由于具有复杂成分的复方中药制剂,在煎煮过程中,其中的某些药物化学成分,受到空气中的氧、热、水的影响,不同组分之间产生了化学变化及相互作用,其生物活性与物理性质发生改变[28],并且由于中药复方的配伍不仅只是药物数量上的简单相加,而是通过药物之间的相互配伍发生了质的改变[29]。这些均导致含有抑菌作用连翘、金银花的上感合剂仅使用简单的培养基稀释法就能有效的将供试品的抑菌作用消除。因此,培养基稀释法对供试品进行处理可满足该制剂对需氧菌、霉菌和酵母菌总数进行计数的需要。
按照微生物限度检查方法验证试验,可将验证菌株分别于样品环节、供试液制备环节及最后环节进行加入,进行回收率测定[30]。综合考虑实验方案的可行性,以及样品与操作过程的影响因素,最合理的加菌方式为供试液制备阶段加菌[30]。本试验优先选择文献报道[30]的在供试液制备环节加菌,通过表1、2试验结果可知,5种试验菌株的回收率均在0.5~2范围内[22],满足药典对验证试验的微生物回收率规定,因此采用供试液制备环节加入试验菌是合理且可行的。通过表1、2试验结果可知,采用平皿倾注法对药典中规定验证用的5种试验菌株回收率均在0.5~2范围内[22],满足药典对验证试验的微生物回收率规定,因此,采用平皿倾注法对微生物进行回收方法准确有效且操作简便。通过表3、4试验结果可知,采用平皿法对控制菌进行检查,试验组中菌生长良好,阴性对照组未检出。药典对控制菌检查方法验证需要设立供试品对照组并没有进行明确规定,本试验通过设立供试品对照组为了证明供试品是否被污染,保证结果的可靠性,如表3结果显示:供试品对照组未检出控制菌,既证实了供试品未被污染,也证明了此方法的可靠性及可行性,符合药典的规定。供试液从制备到加入培养基的时间若超过1 h,即有可能导致微生物的大量繁殖或者死亡,从而影响微生物计数的结果。同时,若制备供试液需水浴加热及培养基需保温,温度均不应超过45℃,且对供试液进行水浴加热的时间也不应超过30 min,避免长时间加热对结果产生影响。
综上所述,上感合剂微生物限度检查方法验证试验符合2015年版《中国药典》四部通则要求,建立的方法准确可行,可用于上感合剂的微生物限度检查,同时,可以为医院口服中药制剂的微生物限度检查方法建立提供参考[8-9,11]。
猜你喜欢
中国测试(2022年4期)2022-05-10
现代仪器与医疗(2021年6期)2022-01-18
食品工程(2020年3期)2020-01-05
计算机测量与控制(2017年6期)2017-07-01
中华灾害救援医学(2015年7期)2016-01-07
现代检验医学杂志(2015年2期)2015-02-06
护理研究(2014年26期)2014-08-15
现代检验医学杂志(2014年1期)2014-02-06
现代检验医学杂志(2014年4期)2014-02-02
科技传播(2011年18期)2011-08-15
