
电感耦合等离子体串联质谱测定高纯稀土及稀土氧化物中超痕量铁
2023-10-08 03:02:44刘宏伟陈雪峰黎树春符靓
分析化学 2023年9期
刘宏伟 陈雪峰 黎树春 符靓
1(湖南工学院材料科学与工程学院, 衡阳 421002) 2(重庆大学材料科学与工程学院, 重庆 400045)
3(重庆上甲电子股份有限公司, 涪陵 408100)
稀土因其独特的光学和磁学性能,广泛用于激光发生、催化反应、能源转换、荧光发光、大功率永磁体以及特种玻璃等先进材料领域[1-3]。稀土中的杂质元素会降低材料的性能,已成为影响稀土应用的关键因素[4],为确保终端产品的高性能,需采用纯度极高的稀土为原材料,严格控制稀土原材料中杂质元素的含量。然而,在高纯稀土的制备过程中会不可避免地引入杂质元素,尤其是铁(Fe),因其在自然界中分布很广[5],已成为高纯稀土中重点监控的杂质元素之一。
根据稀土应用领域的不同,需要使用5N(99.999%)至6N(99.9999%)甚至更高纯度的高纯稀土,即使是5N 的高纯稀土,其杂质总含量也仅为10 mg/kg,单个杂质元素的含量极低,因此,高纯稀土中杂质元素的分析方法要求具有高灵敏度和低检出限。国标法采用分光光度法和电感耦合等离子体发射光谱(ICP-OES)法测定稀土及其氧化物中的Fe[6],虽然分光光度法操作简单、分析成本低,但准确度低、检出限高;ICP-OES 法的灵敏度和准确度高,但光谱干扰严重,检出限偏高,两种分析方法均难以满足4N以上高纯稀土中杂质元素Fe 的检测要求。辉光放电质谱(GD-MS)采用固体导电材料进样技术,可用于高纯物质中痕量以及超痕量杂质元素的分析[7],但需要使用包含分析元素的高纯校准标样,限制了GD-MS 的应用。电感耦合等离子体质谱(ICP-MS)法广泛用于高纯稀土及其氧化物的质量控制,但对于超痕量Fe 的测定,高丰度同位素56Fe 不仅受到基于ICP-MS 氩气形成40Ar16O 的高强度干扰,还会受到来自稀土基质及共存杂质元素所形成的复杂干扰[8]。冷等离子体技术通过抑制生成40Ar16O 离子消除对56Fe 的干扰[9],然而,低温冷等离子体分解基质的能力差,降低了ICP-MS 对高浓度稀土基质的耐受性,影响分析的稳定性和准确性。碰撞/反应池(CRC)为ICP-MS 消除质谱干扰提供了通用方法[10-12],但碰撞模式仅能消除多原子离子干扰,在碰撞过程中分析元素的能量也会受到损失,尤其是痕量元素,其分析受损更严重,从而影响分析灵敏度;而在反应模式下,由于众多不可预知反应的发生,反应产物离子极易对分析元素形成新的质谱干扰,为更好地发挥反应模式消除干扰的能力,必须提高CRC 中离子/分子反应的选择性。
为减少干扰离子进入CRC,研究者在电感耦合等离子体串联质谱(ICP-MS/MS)的CRC 和等离子体之间配置四极杆质量过滤器(Q1),通过Q1阻止大量干扰离子进入CRC,提高CRC 内离子/分子反应的选择性,然后利用另一个四极杆质量过滤器(Q2)控制反应产物离子,从而确保了反应模式的一致性和可靠性[13-16]。在ICP-MS/MS 的串联质谱(MS/MS)模式下,采用O2为反应气,56Fe+与O2的质量转移反应为吸热过程,即使通过调整CRC 参数使56Fe+被加速,增大56Fe+的动能,提高反应效率[17],但生成产物离子56Fe16O+的丰度仍然偏低,不利于超痕量Fe 的测定。采用NH3/He(He 为缓冲气)为反应气,虽然可以采用产物离子56Fe(14NH3)2+为分析离子消除干扰[18],但56Fe+与NH3/He 的反应效率低,仅少量56Fe+能与NH3/He 发生质量转移反应,导致Fe 的分析灵敏度低。N2O 作为高效反应气体,由于破坏N2O 的键能(1.6 eV)低于O2的键能(5.1 eV), N2O 比O2更易发生氧原子转移反应[19],同时,N2O 还能与少量原子离子发生氮原子转移反应和电荷转移反应,提高了N2O 反应模式消除质谱干扰的选择性[20-22]。本研究采用ICP-MS/MS 测定高纯稀土及其氧化物中超痕量Fe,在MS/MS 模式下以N2O 为反应气消除干扰,研究Fe+与N2O 的氧原子转移反应机制,以期为高纯稀土及其氧化物中超痕量Fe 的高灵敏精准测定提供新的分析方法。
1 实验部分
1.1 仪器与试剂
8800 型电感耦合等离子体串联质谱仪(美国Agilent 公司),配备八极杆反应池系统(ORS),#100 标准配置。1000 mg/L Fe 标准溶液、65%(m/m)HNO3、35%(m/m)H2O2(德国Merck 公司);100 mg/L 的Sc、Ge、Rh、In、Tb 和Bi 混合内标溶液(5188-6525,美国Agilent 公司);稀土矿石成分分析标准物质(GBW07158、GBW07159、GBW07160 和GBW07161,辽宁省地质矿产研究院);高纯稀土及其稀土氧化物样品购自中国不同生产厂家。HNO3使用前进一步亚沸蒸馏除杂。
ICP-MS/MS 的操作条件见表1。
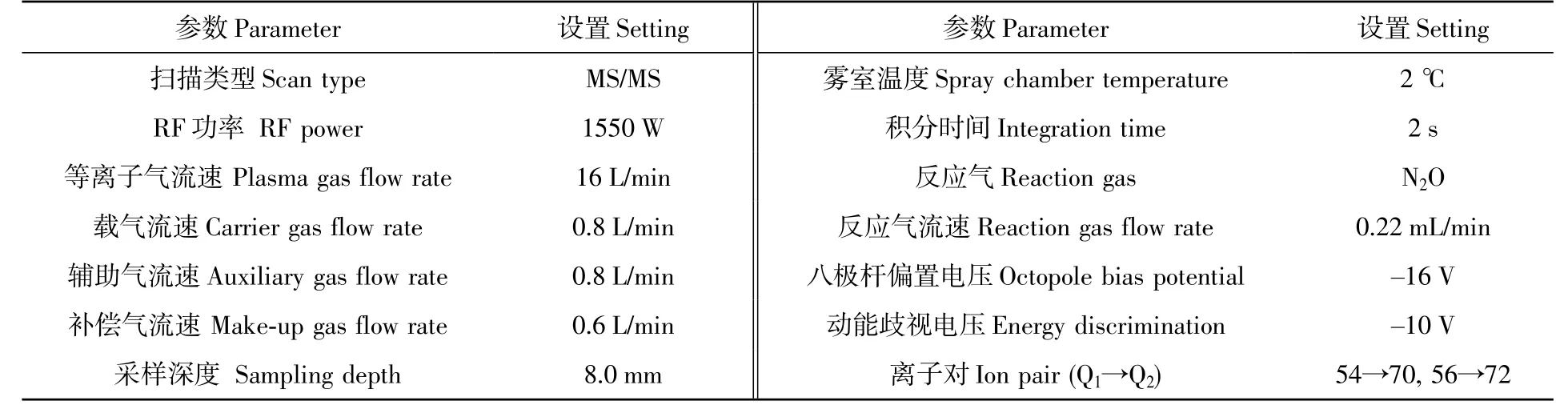
表1 电感耦合等离子体串联质谱(ICP-MS/MS)的操作条件Table 1 Operating conditions of inductively coupled plasmon-tandem mass spectrometry (ICP-MS/MS)
1.2 实验方法
准确称取0.05 g 样品于50 mL 烧杯中,加入少量超纯水润湿,依次加入2.5 mL HNO3和2.5 mL H2O2,低温加热至样品完全溶解,用超纯水定容至50 mL, 制得样品溶液(若样品溶液浓度超出Fe 的线性范围,测定前稀释至Fe 的线性范围内)。随同样品制得空白溶液。
使用热等离子体条件确保ICP-MS/MS 在高浓度稀土基质中具有良好的耐受性。为获得低检出限和高灵敏度,样品分析前采用1 μg/L 的Li、Y、Ce、Tl 调谐溶液对ICP-MS/MS 的测试参数进行优化,使氧化物形成CeO+/Ce+<1%,双电荷形成Ce++/Ce+<3%。为补偿空白溶液、校准标样和样品溶液之间的信号差异,所有溶液中均加入1 mg/L 混合内标溶液。
采用Fe 标准溶液分别配制0.0、1.0、5.0、20 和100 μg/L 系列校准溶液,按ICP-MS/MS 操作参数,依次对校准溶液、空白溶液和样品溶液进行测定,并在线加入1 mg/L 的混合内标溶液,以Fe 与内标元素的相对信号强度所对应的校准溶液浓度建立校准曲线,从而得到样品溶液中Fe 的含量。在MS/MS 模式下,以N2O 为反应气,测定Fe 的工作原理如图1 所示。
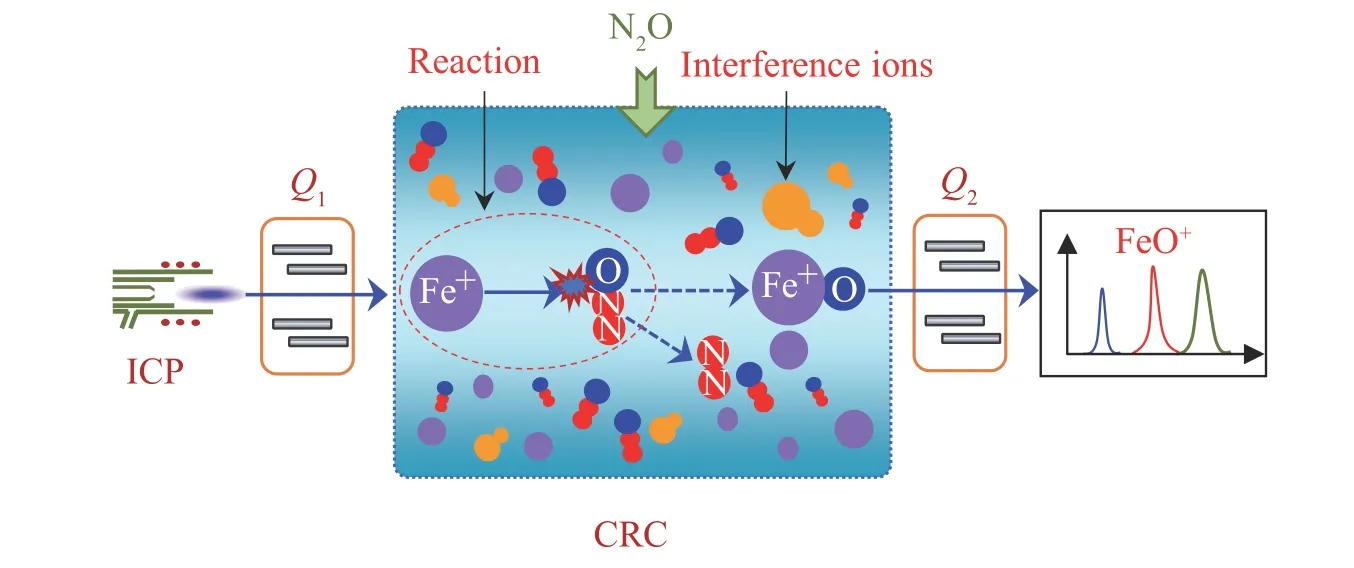
图1 选择N2O 为反应气采用ICP-MS/MS 测定Fe 的原理图Fig.1 Schematic of determining Fe by ICP-MS/MS using N2O as reaction gas
2 结果与讨论
2.1 N2O反应模式下Fe+的质谱行为
Fe 有4 种同位素(54Fe、56Fe、57Fe 和58Fe),所有同位素均受到来自Ar 基离子的严重干扰,同时还受到其它离子的潜在干扰,其中,56Fe 的丰度最高(91.68%),54Fe 次之(5.82%),而57Fe 和58Fe 的丰度低于5%,本研究不予考虑。分别选择54Fe 和56Fe 为分析同位素,在MS/MS 模式下,考察54Fe+和56Fe+在ORS 中采用N2O 为反应气的质谱行为。
54Fe+主要受到来自Ar、Ca 和Cl 形成的质谱干扰(40Ar14N+、40Ca14N+、38Ar15NH+、36Ar18O+、38Ar16O+、36Ar17OH+和37Cl16OH+),由于Cd 和Pd 具有较高的第二电离能(分别为16.85 和19.38 eV), 在等离子体中形成的108Cd++和108Pd++对54Fe+的干扰可以忽略不计。在MS/MS 模式下,设置Q1的质荷比(m/z)为54,54Fe+和m/z54 的干扰离子进入ORS,54Fe+与N2O 发生氧原子转移反应生成了大量54Fe16O+,而所有干扰离子均未与N2O 发生氧原子转移反应(图2A),设置Q2的m/z=70,利用质量转移产物54Fe16O+为检测离子,能彻底消除干扰。56Fe+主要受到来自40Ar16O+、40Ca16O+、40Ar15NH+、38Ar18O+和38Ar17OH+的质谱干扰,而Sn 的第二电离能较低(14.59 eV), 在等离子体中形成少量112Sn++,干扰56Fe+的测定。在N2O 反应模式下,56Fe+的质谱行为与54Fe+相似,与N2O 发生氧原子转移反应生成了大量56Fe16O+(图2B),而干扰离子不与N2O 反应,设置Q1和Q2的m/z分别为56 和72,利用56Fe+→56Fe16O+离子对通过质量转移法消除干扰。
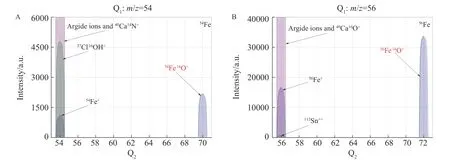
图2 在N2O 反应模式下,(A)54Fe 和(B)56Fe 的质谱扫描图,1 μg/L Fe 中加入1 mg/L Ca、Cl、Cd、Pd和Sn 作为干扰元素Fig.2 Mass spectra scanning diagram of(A) 54Fe and(B) 56Fe in the N2O reaction mode.1 mg/L Ca,Cl,Cd,Pd,and Sn are added to 1 μg/L Fe as interference elements
为评价以N2O 为反应气测定Fe 的方法的有效性,对比分析了不同反应模式下54Fe 和56Fe 的灵敏度和背景等效浓度(BEC),结果见表2。54Fe 和56Fe 在N2O 反应模式下获得的灵敏度最高,O2反应模式次之,在NH3/He 反应模式获得的灵敏度最低,这是由于Fe+与NH3/He 发生质量转移反应,生成的团簇离子众多,影响每种产物离子的丰度,而Fe+与N2O 和O2发生质量转移反应仅生成FeO+,并且N2O 的键能比O2低,发生氧原子转移反应的效率比O2高。54Fe 和56Fe 在N2O 反应模式下的BEC 低于NH3/He 反应模式,略低于O2反应模式,可能是干扰离子在NH3/He 反应模式下形成的少量复杂团簇离子仍然影响了Fe+的测定,采用氧原子质量转移反应(N2O 反应模式和O2反应模式)获得54Fe 和56Fe 的BEC 非常接近,表明质谱干扰已经被消除至背景水平。因此,本研究在MS/MS 模式下选择N2O 为反应气,不仅提高了Fe 的分析灵敏度,而且能彻底消除Fe 受到的质谱干扰,获得极低的BEC。
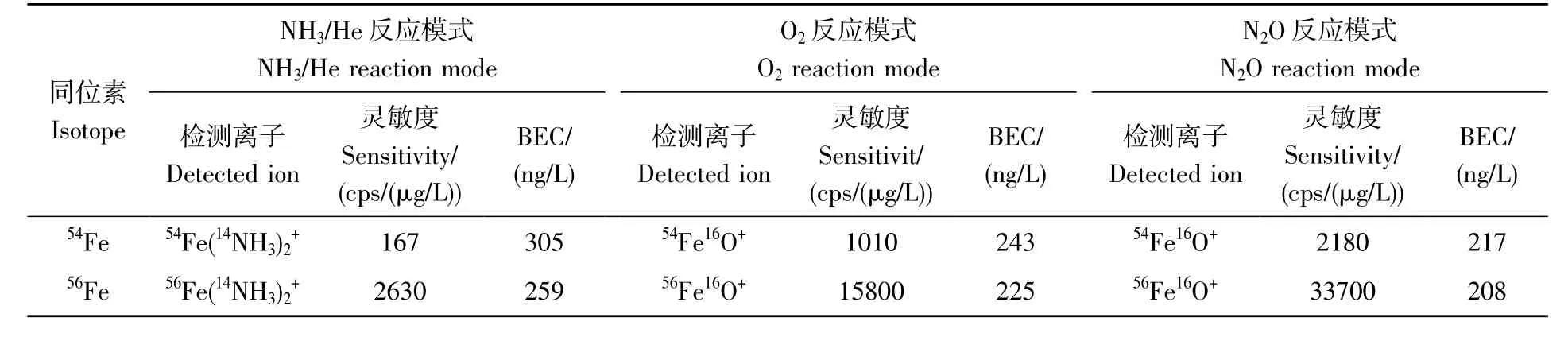
表2 不同反应模式下Fe的灵敏度和背景等效浓度(BEC)Table 2 Sensitivity and background equivalent concentration (BEC) of Fe in different reaction modes
2.2 ORS中Fe+与N2O的反应机制
Fe+在ORS 中与N2O 的反应为:
反应焓ΔHr=D(N2O) -D(Fe+-O),其中,D(N2O)为N2O 的键能(约为1.6 eV),D(Fe+-O)为FeO+的键能(约为3.53 eV), 计算得到Fe+与N2O 的ΔHr= -1.93 eV, 是放热过程,反应能自发进行。为加快反应的进程,实验调整ORS 偏置电压(Voct)为负值,在Fe+与N2O 反应前为其提供质心碰撞能(Ecm)。Ecm与Voct的关系式为[23]:
其中,m(N2O)、m(Fe)、m(Ar)分别为N2O、Fe+和Ar 原子的质量,Vp为等离子体势能(约2 eV),T0为等离子体气体温度(约5000 K),k为Boltzmann 常数。
由式(2)可知,Voct负电压越大,Fe+在反应前获得的Ecm越大,越有利于加速反应进程,单位时间内FeO+的产率越大,分析灵敏度越高,但过大的Voct负电压会导致FeO+的BEC 增大,不利于Fe 的测定。
考察了Voct负电压变化对FeO+的BEC 影响,由图3 可见,54Fe16O+和56Fe16O+的BEC 随Voct的变化趋势基本一致,随着Voct负电压的增大,FeO+的BEC 逐渐变小,当Voct为-16 V 时,54Fe16O+和56Fe16O+的BEC最小,随后继续增大Voct的负电压,54Fe16O+和56Fe16O+的BEC 开始增大。本研究选择Voct=-16 V,依据式(2)计算得到Fe+的Ecm为8.58 eV。在O2反应模式下,设置Voct=-16 V,参照文献[15]计算得到Fe+的Ecm为7.09 eV。这表明在相同Voct下,Fe+在N2O 反应模式下获得的Ecm比O2反应模式大,增加的动能有利于加速反应的进程,而Fe+与O2的反应为吸热过程,增加动能仅促进了吸热反应的发生。实验结果与理论计算一致,表明在N2O 反应模式下,Fe+在N2O 发生了高效氧原子转移反应,与O2反应模式相比,显著提高了分析灵敏度。

图3 八极杆偏置电压(Voct)对FeO+的BEC 影响Fig.3 Effect of octopole bias potential (Voct) on BEC of FeO+
动能歧视电压是碰撞模式下区分分析离子与干扰离子的重要参数,在反应模式下,动能歧视电压需设置为负电压,从而使更多的分析离子进入检测器从而提高分析灵敏度,但动能歧视负电压过高,则会导致分析离子的背景等效浓度(BEC)变大,其优化过程与Voct相似。综合考虑,本实验设置动能歧视电压的最优值为-10 V。
采用空白溶液和1 μg/L Fe 标准溶液优化N2O 流速,考察了不同N2O 流速对背景和56Fe16O+信号强度的影响(见图4),折中选取56Fe16O+高信号强度和低背景所对应的N2O 流速。从图中可以看出,N2O 流速的变化对背景信号强度几乎无影响,背景信号强度始终处于极低水平且保持稳定状态,而随着N2O 流速的增大,56Fe16O+的信号强度先快速增大,当N2O 流速达到0.22 mL/min 时,56Fe16O+信号强度最大;随后增大N2O 流速,56Fe16O+的信号强度开始缓慢变小。因此,本研究选择N2O 的最佳流速为0.22 mL/min。
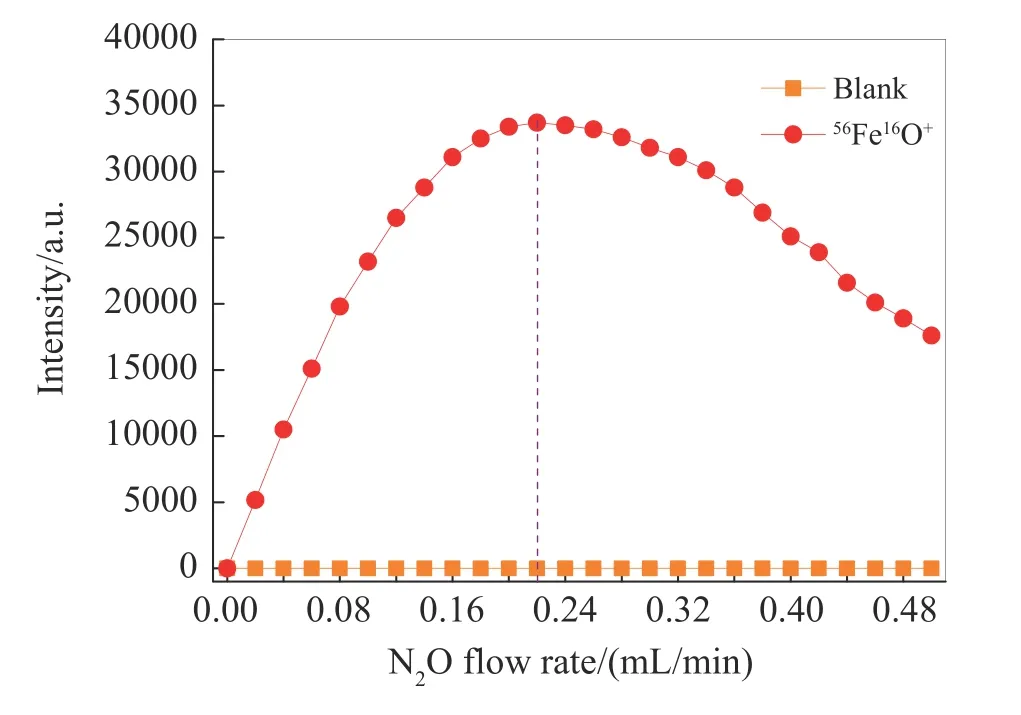
图4 N2O 流速对对56Fe16O+信号强度的影响Fig.4 Effect of N2O flow rate on the signal intensity of 56Fe16O+
2.3 内标离子的优化选择
在ICP-MS 分析中,基于质量数和电离电位与分析元素相近的内标选择原则,通常采用Sc、Ge、Tb、Rh、In 和Bi 作为Fe 的内标元素。然而,在ICP-MS/MS 分析中,由于采用了Fe+的质量转移产物FeO+为检测离子,使内标元素的选择变得复杂[24]。在N2O 反应模式下,内标元素Sc、Ge 和Tb 能与N2O发生高效氧原子转移反应,而Rh、In 和Bi 几乎不与N2O 反应,因此,分别选择45Sc16O+、72Ge16O+、159Tb16O+、103Rh+、115In+和209Bi+为内标离子,通过在60 min 内重复测定样品溶液中加标1 μg/L 的Fe(每间隔10 min 测定1 次,共6 次),考察不同内标离子对测定结果的影响,结果见图5。采用质量转移产物离子45Sc16O+和72Ge16O+所获得的加标回收率明显优于其余4 种内标离子,表明在ICP-MS/MS 分析中,与分析离子质量数相近且具有相同质谱行为的离子更适合于用作内标离子。由于45Sc16O+的灵敏度比72Ge16O+高很多,本实验选择45Sc16O+为内标离子。
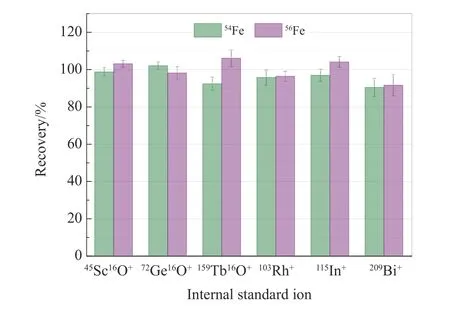
图5 不同内标离子对1 μg/L Fe 加标回收率的影响Fig.5 Effects of different internal standard ions on spiking recovery of 1 μg/L Fe
2.4 方法的分析性能评价
以56Fe 为分析同位素,在选定的ICP-MS/MS 操作条件下,通过测定系列Fe 校准溶液建立校准曲线;以空白溶液11 次重复测定的3 倍标准偏差(3σ)所对应的浓度为Fe 的检出限(LOD)。结果表明,Fe 在0.44~100 μg/L 范围内的线性相关系数为1.0000,线性关系良好,Fe 的LOD 为131 ng/L, 经稀释校正换算方法的检出限(MDL)为131 μg/kg, 明显低于以往类似研究文献报道的LOD[17-18,25]。
由于缺少高纯稀土及其氧化物国家标准参考物质,采用稀土矿石国家标准参考物质(GBW07158、GBW07159、GBW07160 和GBW07161)对方法的准确性和精度进行评价。每个样品平行制备6 份(选取56Fe 为分析同位素,样品溶液稀释1000 倍),测定结果见表3,Fe 的测定值与标准参考物质的认证值基本一致,相对标准偏差(RSD)为1.7%~2.7%,样品的加标回收率为94.0%~105.0%,表明本分析方法的准确性好、精密度高。
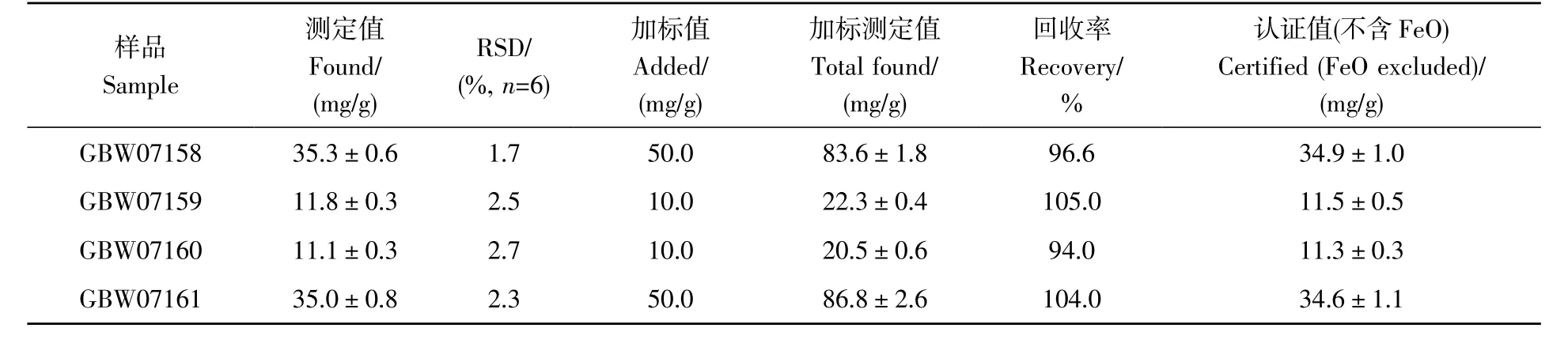
表3 国家标准参考物质中Fe的分析结果Table 3 Analytical results of Fe in national standard reference material
2.5 样品分析
采用本方法分别测定4 种稀土金属和8 种稀土氧化物中的Fe 含量,同时采用国标法(GB/T 12690.5-2017)[6]进行对比分析,每个样品重复测定6 次,结果见表4。4 种稀土金属和8 种稀土氧化物中Fe 的含量存在明显差异,分布在563~5160 μg/kg 之间。采用本方法与国标法的对比分析结果基本一致,说明本方法适用于高纯稀土和稀土氧化物中Fe 的测定。

表4 高纯稀土和稀土氧化物中Fe的分析结果Table 4 Analytical results of Fe in high purity rare earths and rare earth oxides (μg/kg, n=6)
3 结论
提出了基于N2O 为反应气的ICP-MS/MS 消除质谱干扰的新反应体系,其原理是利用N2O 与Fe+发生高效率的O 原子转移反应,通过测定质量转移产物FeO+,消除Fe 的质谱干扰。所建立的分析方法明显优于现有O2反应模式和NH3/He 反应模式的分析效果,具有高灵敏度、低检出限和良好的准确度与精密度,为更多领域复杂体系中超痕量Fe 的检测提供了借鉴。
猜你喜欢
疯狂英语·初中版(2023年5期)2023-06-01 12:31:16
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
食品安全导刊(2021年20期)2021-08-30 06:39:48
中国特种设备安全(2021年12期)2021-04-26 14:37:00
四川冶金(2019年5期)2019-12-23 09:04:36
中成药(2018年6期)2018-07-11 03:01:32
资源节约与环保(2018年1期)2018-02-08 02:18:13
当代化工研究(2016年5期)2016-03-20 16:21:35
中国粮油学报(2016年5期)2016-01-23 02:45:06
机械工程师(2015年10期)2015-02-02 01:14:10
