
基于金属有机骨架DUT-5-2的固相萃取-液相色谱-串联质谱法快速检测生活饮用水中17种全氟烷基物质
2023-10-08 03:02:44吴峰胡月郭萌萌宋志灵谭志军耿倩倩
分析化学 2023年9期
吴峰 胡月 郭萌萌 宋志灵 谭志军 耿倩倩
1(青岛科技大学化学与分子工程学院, 青岛 266042)
2(中国水产科学研究院黄海水产研究所,农业农村部水产品质量安全检测与评价重点实验室, 青岛 266071)
3(青岛农业大学食品科学与工程学院, 青岛 266109)
全氟烷基物质(Perfluoroalkyl substances,PFAS)是一类新型污染物(Emerging contaminants,ECs)。PFAS 由一端的疏水性氟化烷基链和另一端的亲水官能团组成[1],由于C—F 键的键能高,使得这类化合物具有很高的热稳定性和化学稳定性。这种物理和化学性质的独特组合以及疏水和疏油特性,使PFAS非常适合作为防水、防油材料以及抗摩擦剂[2]。因此,PFAS 被大量生产并应用于工业(造纸、纺织品、杀虫剂、石油和矿物和金属电镀等)和消费品(食品包装、化妆品和个人护理产品、油漆、表面活性剂、消防泡沫和防水产品)领域[3-4]。目前,由于PFAS 的广泛使用,其在环境中的浓度相对较高,并且广泛分布于世界各地的地表水、地下水等水体、沉积物和生物体中[5-6]。例如,在骆马湖表层水中共检测出15 种PFAS,∑PFAS 的含量范围为46.09~120.34 ng/L[7];在天津市主要河流中共检测出12 种PFAS,∑PFAS 的含量范围为3.93~357.85 ng/L[8];在珠江水系北江和西江河水中PFAS 的分布和来源的调查[9]中发现,地表水样中共检测出11 种PFAS,旱季时∑PFAS 浓度范围为0.775~441 ng/L, 雨季时为2.66~1060 ng/L;对黄土高原地下水样品中残留的PFAS 进行测定,∑PFAS 的含量范围为2.78~115 ng/L[10]。由此可见,水环境中PFAS 污染现象普遍存在。生活饮用水作为水环境的一部分,是人类接触PFAS 的重要来源之一,已受到越来越严格的管控。2016 年,美国环保局规定饮用水中全氟辛酸(Perfluorooctanoic acid,PFOA)和全氟辛烷磺酸(Perfluorooctanesulfonic acid,PFOS)[11-12]的累计含量不得超过70 ng/L;2020 年12 月,欧洲议会通过修订版《饮用水指令》,明确饮用水中所有PFAS 浓度之和的限值为0.5 μg/L;2022 年3 月,我国发布GB5749-2022《生活饮用水卫生标准》[13],明确规定生活饮用水中PFOA 和PFOS 的限量标准分别为80 和40 ng/L;2022 年12 月,生态环境部发布了重点管控新污染物清单(2023 年版),其中包括PFOA、PFOS、全氟己烷磺酸(Perfluorohexanesulfonic acid,PFHxS)及其盐类和相关化学品。目前,我国尚缺乏对生活饮用水中PFAS 检测的技术标准,因此,开发生活饮用水中PFAS的快速、准确和高灵敏的检测技术是实现上述管控目标的重要基础。
目前,针对生活饮用水等水体中PFAS 的检测方法多采用液相色谱-串联质谱法[14-16]。但是,由于水体样品中干扰物甚多,并且样品中PFAS 通常为痕量水平,难以直接检出,因此需对水样中目标分析物进行选择性富集净化。固相萃取法(Solid phase extraction,SPE)[17]是水体样品最常用的样品前处理技术,其基本原理是利用待测组分在固相填料中的选择性吸附和洗脱,达到分离和富集的目的。SPE 的优点是萃取时间短、有机溶剂使用量少,集萃取和净化为一体。但是,现有的用于水体样品的SPE 法普遍存在SPE 柱成本较高、前处理过程上样量大[15-16]、耗时长、选择性差以及回收率低等缺点[17]。
本研究采用金属有机骨架材料DUT-5-2 通过静电相互作用和疏水相互作用实现对PFAS 的选择性吸附,以此材料为吸附剂,组装得到了能够快速同时富集多组分PFAS 的SPE 柱,通过优化SPE 柱的上样pH 值、洗脱剂用量及再生方法等,建立了固相萃取-超快速液相色谱-串联质谱同时测定生活饮用水中17 种PFAS 的方法,并将其应用于实际生活饮用水样品的检测。本研究为PFAS 污染监控提供了准确、高效且经济的方法。
1 实验部分
1.1 仪器、试剂与材料
Prominence UFLC 液相色谱(日本Shimadzu 公司);5500QTRAP 四极杆-线性离子阱复合质谱(美国AB SCIEX 公司);XW-80A 旋涡混合器(上海医大仪器厂);Himac CR 22GⅡ高速离心机(德国Hitachi公司);N-EVAP 112 氮吹仪(美国Organomation 公司);Flexiwave 型微波合成仪(意大利Milestone 公司);Milli-Q 超纯水仪(美国Millipore 公司);JSM-7500F 型扫描电子显微镜(SEM,日本电子公司);D8 ADVANCE 型X 射线衍射仪(XRD,美国布鲁克公司);Nicolet Nexus IS10 型傅里叶红外光谱仪(FI-IR,美国赛默飞公司)。
17 种标准物质及10 种内标物质,纯度均大于98%,购于加拿大Wellington Labortories 公司,包括(1)外标物质:全氟己酸、全氟庚酸、全氟辛酸、全氟壬酸、全氟癸酸、全氟十一烷酸、全氟十二烷酸、全氟十三烷酸、全氟丁烷磺酸、全氟己烷磺酸、全氟庚烷磺酸、全氟辛烷磺酸、全氟壬烷磺酸、全氟癸烷磺酸、全氟十一烷磺酸、全氟十二烷磺酸、全氟十三烷磺酸;(2)内标物质:13C5-全氟己酸、13C4-全氟庚酸、13C8-全氟辛酸、13C9-全氟壬酸、13C6-全氟癸酸、13C7-全氟十一烷酸、13C2-全氟十二烷酸、13C3-全氟丁烷磺酸、13C3-全氟己烷磺酸、13C8-全氟辛烷磺酸。
甲醇和水(质谱级,美国Merck 公司);乙酸铵(色谱级,美国Sigma Aldrich 公司);氨水(ACS 级,美国Sigma 公司);Oasis HLB 固相萃取柱(6 mL,200 mg, 美国Waters 公司)和Oasis WAX 固相萃取柱(6 mL,150 mg,美国Waters 公司);固相萃取柱空柱套装(6 mL,日照科谱诺公司);Al(NO3)3·9H2O、4,4′-联苯二甲酸(BPDC)和N,N-二甲基甲酰胺(DMF)。其余试剂如未作特殊说明均为分析纯;实验用水为超纯水(18.2 MΩ·cm)。
1.2 SPE柱的组装
首先制备DUT-5-2 金属有机骨架吸附材料。按照Al(NO3)3·9H2O 与BPDC 的物质的量的比为1.3∶1的比例,将0.968 g Al(NO3)3·9H2O 和0.484 g BPDC 溶于60 mL DMF 中,搅拌至完全溶解。将混合溶液转移至聚四氟乙烯的微波反应管中,采用微波合成仪进行微波反应,控制微波功率为200 W,以20 ℃/min的升温速率升至120 ℃,持续反应60 min。冷却至室温后离心分离(4000 r/min,15 min),采用DMF 和甲醇分别洗涤沉淀2~3 次,在80 ℃真空干燥箱中烘干8 h,获得DUT-5-2 金属有机骨架材料。
采用上述合成的DUT-5-2 材料进行SPE 柱的组装。取5 mg DUT-5-2 吸附材料置于双层筛板之间并压实,固定在6 mL SPE 空柱底部,备用。
1.3 SPE步骤
采用6 mL 甲醇和6 mL pH= 3 的纯水活化SPE 柱。取20 mL 待测水样,调节水样的pH=3,加入内标物质(各1 ng), 将水样充分混匀后过SPE 柱,控制流速为每秒1 滴,弃去流出液。用4 mL 甲醇洗脱,收集洗脱液至10 mL PP 离心管中,于室温下氮吹至略低于0.5 mL, 用甲醇定容至1 mL。涡旋混匀30 s, 超声1 min, 以12000 r/min 离心10 min, 取上清液,待测。
1.4 仪器分析
液相色谱条件 Kinetex XB-C18色谱柱(100 mm×2.1 mm,2.6 μm);延迟柱:C18色谱柱(50 mm×2.1 mm,5 μm);柱温:40 ℃;流速:0.30 mL/min;进样量:5 μL;流动相A 为5 mmol/L 乙酸铵溶液,B 为纯甲醇。洗脱梯度:0~1.5 min,10%B;1.6~2.0 min,10%~40% B;2.1~5.0 min, 40%~50% B;5.1~10.5 min,50%~95% B;10.6~14.0 min,95% B;14.1~16.0 min,95%~98% B;16.1~18.0 min,10% B。
质谱条件 电喷雾离子源(ESI),多反应监测(MRM),负离子模式;喷雾电压:-4.5 kV;气帘气压:0.24 MPa;碰撞气压:0.02 MPa;温度650 ℃;碰撞室入口电压:-10 V;碰撞室出口电压:-12 V;驻留时间:20 ms;离子源Gas1: 0.34 MPa; Gas2: 0.34 MPa。化合物的母离子、子离子、解簇电压(DP)、碰撞能(CE)和碰撞室射出电压(CXP)等参数见表1。
1.5 质量控制
为防止样品前处理过程中引入高背景值,实验过程中避免使用聚四氟乙烯材质的器皿,并且器皿在使用前用超纯水和甲醇充分清洗。采用标准曲线校正,同位素内标法进行定量分析。每批样品进行测试时,同时做空白实验、基质加标实验和线性实验,空白实验确保试剂空白中所有分析物的含量低于定量限;基质加标实验加入PFAS 标准物质(0.5、2 和10 ng)和内标物(1 ng)验证方法的准确度,回收率控制在60%~120%范围内;采用至少5 个浓度点进行标准曲线的绘制,并且线性相关系数应达到0.995 以上。
2 结果和讨论
2.1 DUT-5-2材料的形态和结构表征
2.1.1 SEM、XRD和FT-IR分析
采用SEM 对DUT-5-2 材料进行观察。如图1 所示,合成的DUT-5-2 材料呈不规则球形,粒径在500~700 nm 之间,颗粒相对均匀,整体结构呈疏松多孔的典型形貌,此形貌特点有利于在吸附过程中提供更多的吸附位点,以增强对PFAS 的吸附效果。

图1 微波合成的DUT-5-2 的扫描电镜(SEM)图像Fig.1 Scanning electron microscopy (SEM) image of DUT-5-2 synthesized by microwave-assisted method
DUT-5-2 的XRD 结果如图2A 所示,在6.1°、12.1°和18.3°处的特征衍射峰分别与(010)、(020)和(030)晶面相对应,与文献[18]报道的DUT-5(Al)基本一致,说明成功合成了DUT-5-2。图2B 为DUT-5-2及其合成原料(Al(NO3)3·9H2O,BPDC)的FT-IR 光谱图。DUT-5-2 和Al(NO3)3·9H2O 分别在3420 cm-1附近出现O—H 伸缩振动峰,DUT-5-2 与BPDC 在1430~1600 cm-1之间具有相似的苯环骨架振动吸收峰。上述结果进一步表明已成功合成DUT-5-2 材料。

图2 (A)DUT-5-2 的X-射线衍射(XRD)谱图;(B)DUT-5-2 及其原料Al(NO3)3·9H2O 和BPDC 的傅里叶变换红外(FT-IR)光谱图Fig.2 (A)X-ray diffraction(XRD)pattern of DUT-5-2;(B)Fourier transform-infrared(FT-IR)spectra of DUT-5-2,Al(NO3)3·9H2O and BPDC
2.1.2 DUT-5-2的比表面积、孔体积和孔径的测定
DUT-5-2 的氮气吸附-脱附等温线如图3 所示,在低压段,N2吸附量(cm3/g)迅速增加(P/P0=0.0~0.1)。根据国际纯化学与应用化学联合会分类,DUT-5-2 呈现出Ⅰ型等温线,这是微孔材料的典型特征。

图3 DUT-5-2 的氮气吸附-脱附等温线Fig.3 Nitrogen adsorption-desorption isotherms of DUT-5-2
DUT-5-2 材料的微孔性能如表2 所示。DUT-5-2 的BET(Brunauer-Emmett-Teller)模型比表面积为1840 m2/g,孔体积为0.93 cm3/g,孔径为0.85 nm。本研究合成的DUT-5-2 比文献[18-19]报道的DUT-5 具有更高的比表面积,可为目标物质吸附提供更多的吸附位点,从而提高吸附容量。

表2 DUT-5的微孔性能Table 2 Microporous properties of DUT-5
2.2 DUT-5-2吸附剂用量
吸附剂用量是影响SPE 方法回收率的关键因素。吸附剂用量少会导致PFAS 吸附不完全,吸附剂用量过多可能使杂质吸附增加。以自来水为空白水样,加标浓度为2 μg/L, 比较了不同用量(1、5、10 和20 mg)的吸附剂DUT-5-2 对SPE 回收率的影响。实验结果表明,当DUT-5-2 的用量由1 mg 增至5 mg 时,方法回收率由36%提高至98%;当DUT-5-2 的用量超过5 mg 时,方法回收率并未显著增加,维持在90%~100%之间。综合考虑成本和环保,选择DUT-5-2 的最佳用量为5 mg。
2.3 样品前处理条件的优化
2.3.1 样品溶液pH值的影响
样品溶液的pH 值决定了吸附剂在溶液中所带电荷的不同,导致其与PFAS 之间的作用力也不同[20-22],进而影响SPE 对目标物的富集效果。考察了自制SPE 柱在不同pH 值下对17 种PFAS 化合物的富集效果,结果如图4 所示,当样品溶液pH=2 时,17 种目标化合物的平均回收率范围在51.0%~132.0%之间;当样品溶液pH=3 时,17 种目标化合物的平均回收率范围在63.1%~115.5%之间;当样品溶液pH=4、5和8 时,17 种目标化合物的平均回收率最低值不足60%,最高值均超过120%。结合文献[20]可知,当样品溶液pH=3 时,DUT-5-2 吸附剂的表面带正电荷,此时可以通过静电相互作用有效地吸附PFAS;当样品溶液pH>4.83(等电点)时,DUT-5-2 的表面所带电荷变为负电荷,导致与PFAS 之间产生静电排斥,使其吸附效率降低。因此在后续实验中,选择将样品溶液的pH 值调整为3 进行SPE。
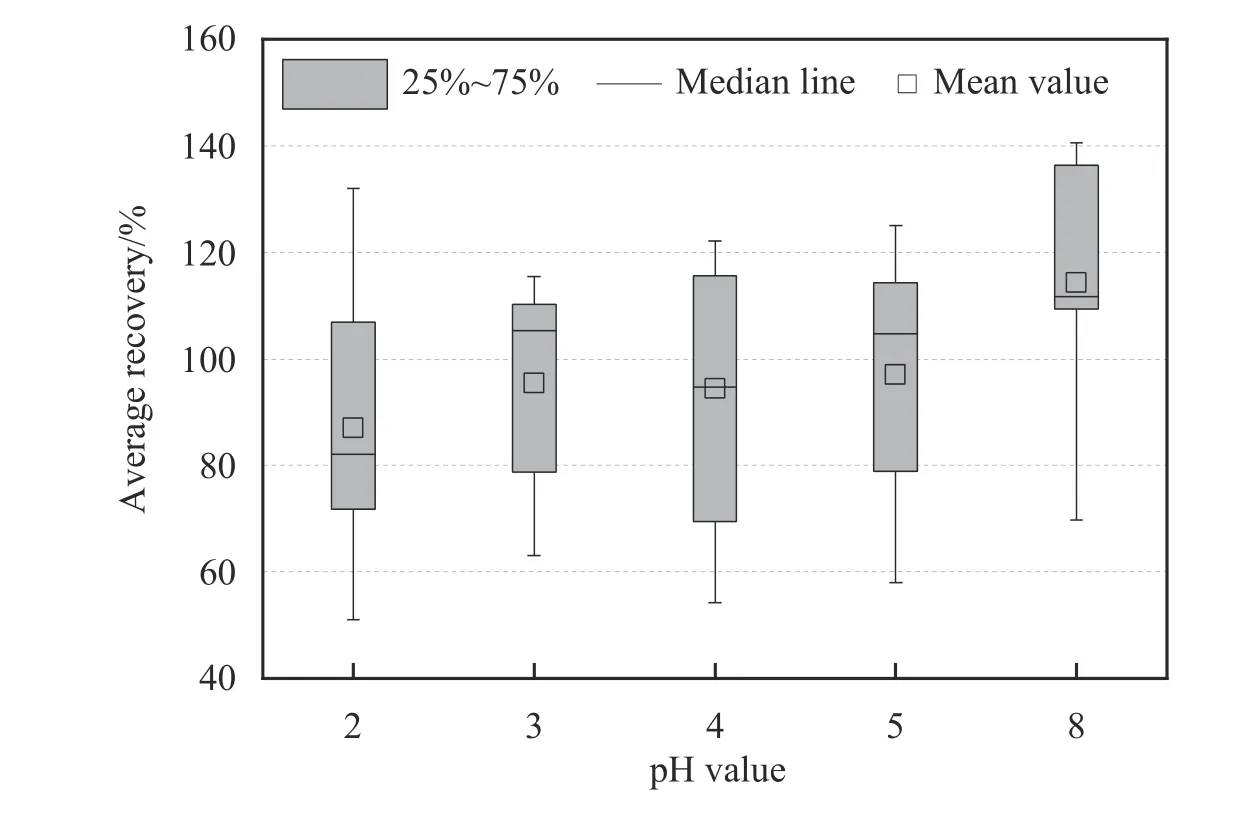
图4 样品溶液的pH 值对PFAS 平均回收率的影响Fig.4 Average recoveries of PFAS at different pH values of samples
2.3.2 洗脱剂种类与用量的影响
不同极性的有机溶剂对化合物的洗脱能力不同,而洗脱剂用量会影响目标物解吸的程度,因此对洗脱剂种类与用量进行了优化。参考文献[20]的方法,选择甲醇、0.1%氨水-甲醇、0.5%氨水-甲醇和1%氨水-甲醇为洗脱剂,重复测定3 次,以平均回收率考察不同溶剂的洗脱效果。当洗脱剂为甲醇时,17 种PFAS 的平均回收率为63.1%~115.5%;当洗脱剂分别为0.1%氨水-甲醇、0.5%氨水-甲醇和1%氨水-甲醇时,有3~4 种目标化合物的平均回收率低于60%,有2~7 种目标化合物的平均回收率高于120%(图5A,M 代表甲醇)。

图5 (A)使用不同洗脱剂时各组分的平均回收率;(B)不同甲醇用量下各组分的平均回收率Fig.5 (A) Average recoveries using different types of eluting solvents; (B) Average recoveries of each component using different volumes of methanol
本实验以简化前处理步骤、减少有机试剂使用量为原则,进一步考察了不同体积的甲醇洗脱剂的影响。实验结果如图5B 所示,当甲醇用量为2 mL 时,PFHxA 的平均回收率超过120%,PFDoS 的平均回收率低于60%;当甲醇用量为4 mL 时,17 种目标化合物的平均回收率在63.1%~115.5%范围内;当甲醇用量为6 mL 时,PFHxA 和PFNS 的平均回收率均高于120%。因此,选择4 mL 甲醇作为洗脱剂,获得了较满意的洗脱效果。
2.4 方法学验证
取适量PFAS 混合标准溶液和内标溶液,配制一系列浓度梯度的标准工作溶液,按照1.3 节的条件进行测定,以各组分和内标物的峰面积比值为纵坐标,各组分的质量浓度为横坐标进行线性回归分析,17 种化合物的线性良好,检出限(S/N=3)为0.8~8.0 ng/L, 定量限(S/N=310)为2.5~25.0 ng/L(表3),远低于生活饮用水中PFAS 的限量要求。

表3 17种PFAS目标物的线性范围、回归方程、相关系数、检出限和定量限Table 3 Linear ranges, regression equations, correlation coefficients, limits of detection (LODs) and limits of quantification(LOQs) of 17 kinds of target PFAS
选用瓶装纯净水、自来水和饮用矿物质水3 种日常生活中的常见生活饮用水[23],分别添加0.5、2.0和10.0 μg/L 的混合标准溶液,每组设6 个平行样,采用本方法进行检测,计算加标回收率和相对标准偏差(RSD),结果见图6。实际加标样品测定的液相色谱-串联质谱色谱图见图7。结果表明,17 种PFAS在瓶装纯净水中的加标回收率为63.9%~116.6%之间,相对标准偏差(RSD)为0.59%~9.19%;17 种PFAS在自来水中的加标回收率为68.9%~119.0%,RSD 为1.1%~13.5%;17 种PFAS 在饮用矿物质水中的加标回收率为62.9%~119.9%,RSD 为0.4%~11.0%。上述结果表明,本方法的准确度和精密度良好,可应用于实际生活饮用水的检测。
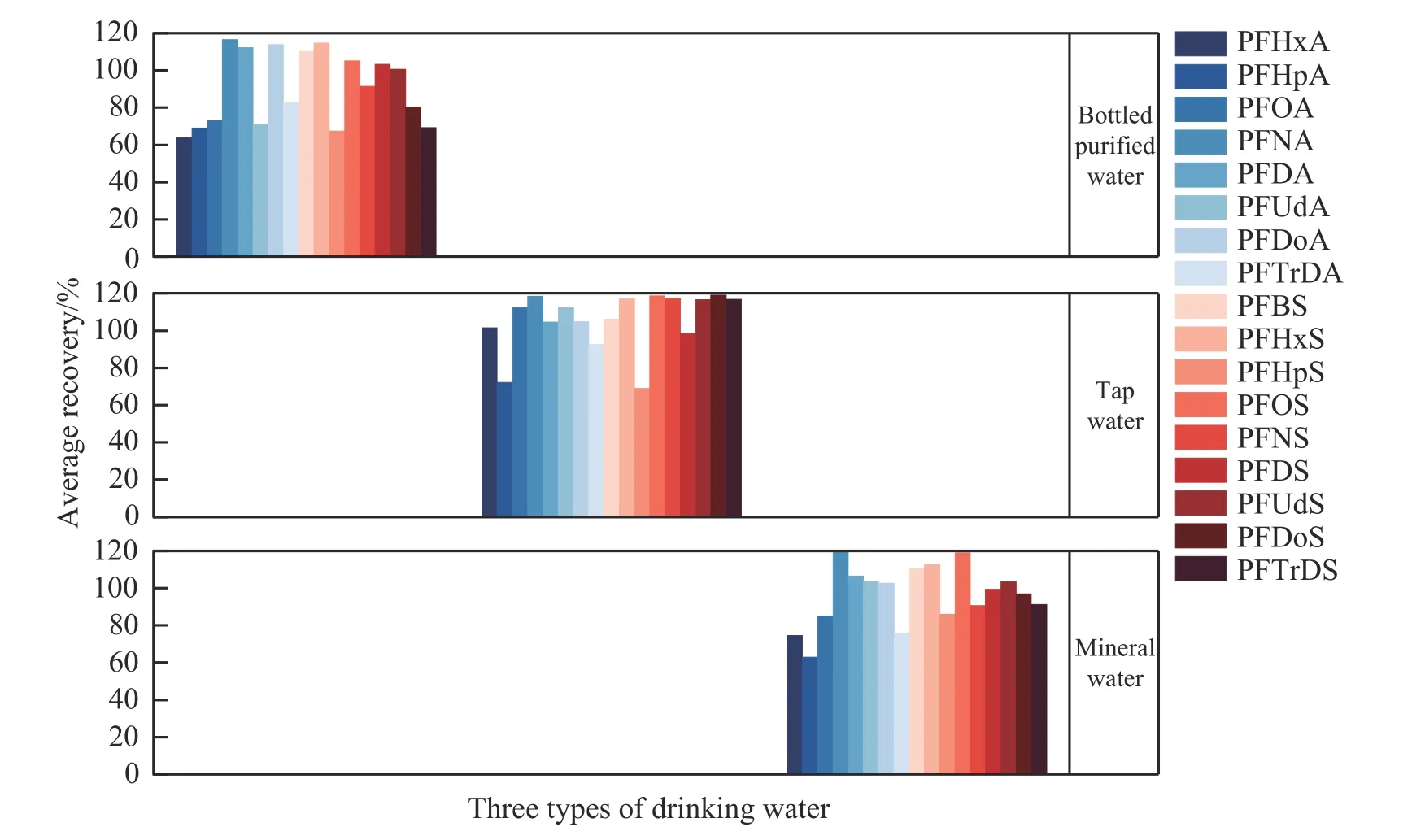
图6 3 种生活饮用水样品中17 种PFAS 的平均回收率Fig.6 Average recoveries of 17 kinds of PFAS in three types of drinking water
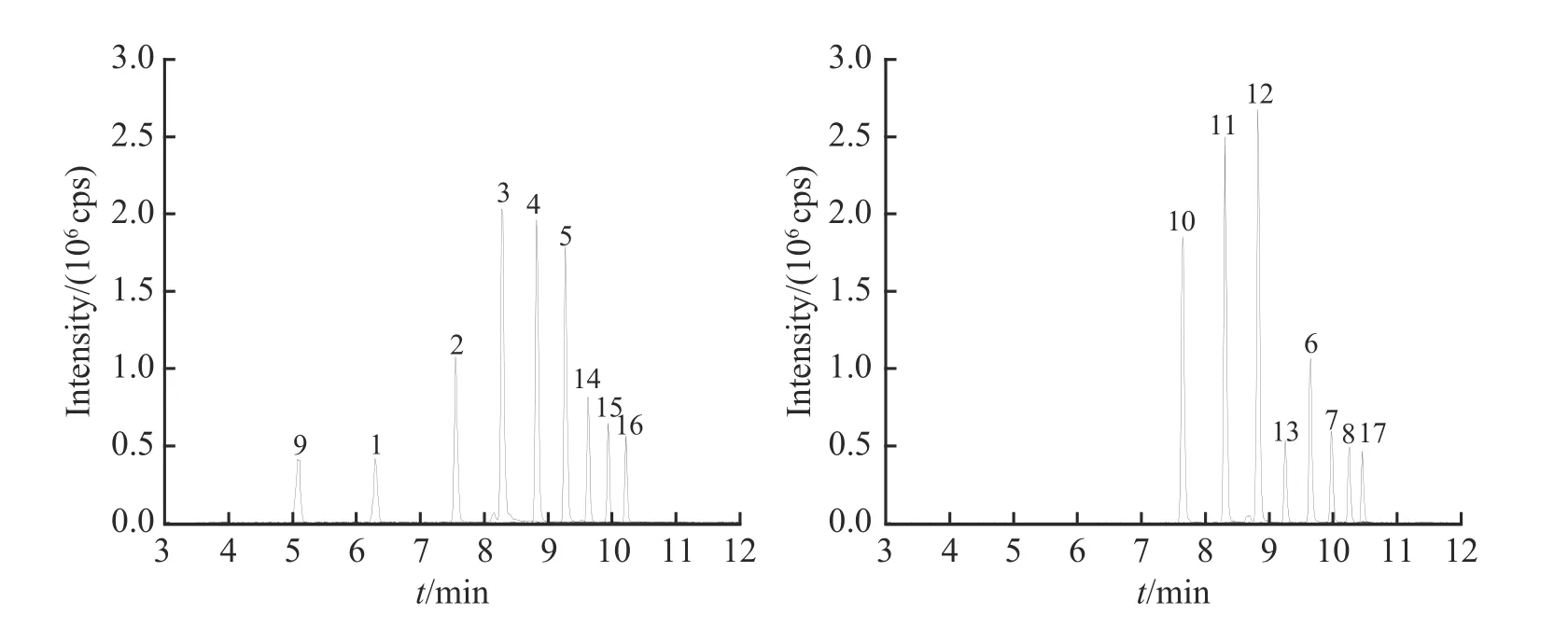
图7 实际加标样品中PFAS(2.0 μg/L)的提取离子色谱图(图中化合物序号与表3 一致)Fig.7 Extracted ion chromatograms of PFAS (2.0 μg/L) in actual spiked samples (The serial numbers of compounds are the same as those in Table 3)
2.5 自制SPE柱与商品化Oasis HLB和Oasis WAX SPE柱的对比
目前,水体中PFAS 的检测方法常使用商品化的SPE 柱进行目标物的富集,如Oasis HLB 和Oasis WAX 均对PFAS 具有较好的富集效果。为探究本研究自制SPE 柱的优势,选用Oasis HLB 和Oasis WAX两种SPE 柱与自制SPE 柱进行比较,采用相同体积的瓶装纯净水作为水样进行加标,考察回收率。自制SPE 柱萃取步骤参照1.3 节,Oasis HLB 和Oasis WAX 的萃取步骤参考文献[24]中的最优条件,平行测定3 次,具体实验条件设置见表4。

表4 对比实验的条件设置Table 4 Condition settings for comparative experiments
实验结果如图8 所示,自制SPE 柱的平均回收率在77.5%~120.0%范围内,RSD 为0.1%~14.8%,相比于Oasis HLB 和Oasis WAX 两种固相萃取柱的平均回收率(13.5%~127.8%和6.7%~120.7%)和RSD(0.1%~15.8%和0.4%~20.4%),其准确度和精密度更高;自制SPE 柱仅需4 mL 甲醇即可完全洗脱目标吸附物质,在15 min 内可完成整个萃取流程,相对于Oasis HLB 固相萃取柱需30 min 和Oasis WAX 固相萃取柱需40 min,其前处理步骤更加快速;以原料成本进行测算,自制SPE 柱的成本约为5 元/样,远低于Oasis HLB(约60 元/样)和Oasis WAX(约70 元/样)两种SPE 柱,具有价格低廉的优势。综上所述,本实验所使用的固相萃取柱较常用的Oasis HLB 和Oasis WAX 商品化SPE 柱,具有快速、经济、准确度高和精密度好的优势。

图8 3 种SPE 柱的平均回收率Fig.8 Average recoveries of PFAS using three kinds of SPE columns
2.6 SPE柱的再生
以节约成本和环保为原则,对使用后的SPE 柱进行回收再生实验。采用25 mL 甲醇于25 ℃、150 r/min的恒温水浴振荡中对吸附剂进行解吸,再用25 mL 超纯水洗涤3 次,真空干燥,获得再生一次的材料。使用再生后的材料重新组装SPE 柱,并对其进行加标回收实验。实验结果表明,再生后的SPE 柱仍能有效吸附17 种目标化合物,并且经过4 次循环后,回收率在62.6%~119.2%范围内,这表明自制SPE 柱具有良好的再生性能。
2.7 实际饮用水分析
应用本方法测定了18 份实际生活饮用水样品(自来水、饮用矿物质水和瓶装纯净水各6 份),发现部分样品存在PFAS 污染,总量为9~394 ng/L(表5);17 种PFAS 在瓶装饮用水样品中均未检出;PFOA 和PFOS 在所有样品中均未检出。在2 份样品中均检出重点管控新污染物PFHxS,因此建议继续开展对生活饮用水中PFAS 的污染监测工作。

表5 阳性水样中PFAS的检测结果Table 5 Concentrations of PFAS in contaminated drinking water samples
3 结论
以金属有机骨架材料DUT-5-2 为吸附剂,组装SPE 柱并进行SPE,实现了生活饮用水中17 种PFAS的同步富集和净化,采用液相色谱-串联四极杆/线性离子阱复合质谱进行测定,建立了一种快速检测生活饮用水中17 种PFAS 的分析方法。本方法的上样体积仅为20 mL, 省去了常规SPE 净化方法的淋洗步骤,有效缩短了整个萃取流程的时间。本方法的灵敏度满足生活饮用水中PFAS 的限量要求,准确度和精密度良好,适用于实际水样的检测。自制的SPE 柱价格低廉、可再生,并且可拓展用于更多PFAS 组分的检测,具有良好的应用前景。本方法可改善我国实施PFAS 管控以来对生活饮用水中PFAS 检测方法匮乏的现状,为生活饮用水中PFAS 污染监测提供了经济、高效和可靠的分析手段。
猜你喜欢
中国环境科学(2023年9期)2023-09-23 12:10:20
供水技术(2022年1期)2022-04-19 14:11:42
化工生产与技术(2016年5期)2016-03-13 10:07:26
化工生产与技术(2016年5期)2016-03-13 10:07:26
食品界(2016年4期)2016-02-27 07:36:15
中国生化药物杂志(2015年4期)2015-07-07 12:05:44
湖南师范大学自然科学学报(2015年2期)2015-02-27 14:50:13
中国药业(2014年21期)2014-05-26 08:56:50
化工环保(2014年6期)2014-04-04 10:53:28
郑州大学学报(理学版)(2014年4期)2014-03-01 04:21:19
