
Mazabraud 综合征的临床、影像学及病理学特征:1 例报道暨文献复习
2023-10-07 13:13:52沈艳杨通陈贵东古婉仪
肿瘤预防与治疗 2023年9期
沈艳,杨通,陈贵东,古婉仪
510260 广州,广州医科大学附属第二医院 病理科
Mazabraud 综合征是以单骨性或多骨性的纤维结构不良(fibrous dysplasia,FD)合并单发或多发性肌内黏液瘤(intramuscular myxoma,IM)为临床特征的罕见疾病[1],发病率约1/1 000 000(www.orpha.net)。由于该疾病常缺乏明显的临床症状,并且临床工作者认识不足易导致漏诊,预计其发病率可能高于预期。 鉴于骨病变恶性转化的风险增加,有必要提高对Mazabraud 综合征的认识,避免误诊、漏诊。现报道我院收治的1 例Mazabraud 综合征,并复习相关文献,探讨其病因、临床特点和病理学特征,以期加深临床医师对该疾病的了解。
1 病例报道
患者,女性,57 岁,因左臀部肿物3 年余伴左髋部疼痛1 月,于2021 年6 月入院。自述平素身体良好,否认既往有外伤史或肿瘤史。入院后彩超显示左大腿根部内侧皮下约3.8 mm 处脂肪层大小约66 mm× 23 mm× 39 mm 混合回声团块,边界尚清晰,形态欠规则,内部回声不均,可见液性暗区,后方回声增强。X 线检查显示左侧髂骨、左侧股骨上段骨质密度不均匀,见斑片状低密度影及稍高密度影(图1A)。MRI 示左侧股骨上段、髋臼、坐骨、髂骨见多发片状异常信号,为T1WI 低信号混杂点状高信号,T2WI 及压脂序列混杂高信号,DWI 高信号,增强扫描可见不均匀强化(图1B)。左侧大腿内侧软组织见肿块影,范围约4.2 cm×2.8 cm,为T1WI 低信号、T2WI 高信号、压脂序列高信号,增强扫描为不均匀增强,边缘见环形强化(图1C、D)。
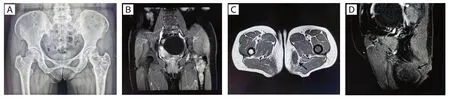
图1 本病例的影像学表现Figure 1.Imaging Features of Present Case
完善相关检查后,行左臀部肿物切除术及左股骨近端骨病灶刮除及植骨术。切除和刮除组织分别送病理检查。大体观察见:(1)左臀部肿物,大体为椭圆形,表面光滑,似有包膜,附着少量脂肪,大小5.5 cm×4.0 cm ×2.8 cm,切面呈实性,灰白色,质软,有光泽,呈胶冻样(图2);(2)左股骨近端骨病灶刮除标本:碎组织一堆,约4.5 cm×4 cm×1.5 cm,灰白色,质较韧,刀切有砂砾感。镜下观察见:(1)左臀部肿物:肿瘤细胞稀疏,间质富含黏液样基质,可见疏松的网状纤维,血管稀少(图3A)。细胞小且大小较一致,胞浆少,嗜酸性,呈梭形或星芒状,核呈圆形或梭形,固缩深染,无异型性,无核分裂像(图3B)。肿瘤周围可见不完整的假性纤维包膜包绕,可见肿瘤细胞侵入脂肪间,与周边肪组织分界不清(图3C);(2)左股骨近端病灶:由增生的纤维样组织和不成熟的编织状新生骨构成,两者比例在不同区域多少不等。纤维样组织主要由纤维母细胞样梭形细胞构成,形态温和,无明显异型性,核分裂少见;编织状新生骨主要为纤细、形态不规则、弯曲的的骨小梁,呈“C””J”等英文字母形,骨小梁之间缺乏彼此连接,表面未见明显骨母细胞衬覆(图3D)。局部间质见黏液样变及散在或灶性泡沫样组织细胞浸润。免疫组织化学染色发现,左臀部肿瘤细胞呈vimentin 阳性,Desmin、S-100、SMA、CD34、MUC4 均阴性。结合相关结果,本病例最终诊断为:Mazabraud 综合征(左股骨近端FD 合并左臀部IM)。患者术后门诊随访1 年10 个月,未见复发或恶性转化。

图2 软组织肿物大体标本Figure 2.Gross Specimen of Soft Tissue Tumor

图3 本病例的病理学特征Figure 3.Pathological Features of Present Case
2 讨 论
1926 年Henschen[1]首次报道了1 例FD 同时伴发周围多发性IM 的病例,1957 年Mazabraud 进一步详细阐述了FD 与IM 之间的病理相关性,并以自己的名字将其命名为Mazabraud 综合征[2]。这是一种罕见的散发性疾病,病因尚不明确。近年来,分子遗传学研究显示FD 和IM 都与GNAS1 基因突变有关[3-4],且突变热点几乎均位于第8 外显子,提示GNAS1 基因突变在Mazabraud 综合征发病中起着决定性作用。人G 蛋白分为激活型(Gs)和抑制型(Gi)两种,由α、β、γ亚基组成,在细胞跨膜信号传递中起重要作用。α亚基是其活性亚基,含有GTP和 GDP 结合位点并具有GTP 酶活性[5]。GNAS1 基因突变可导致其编码蛋白Gsa 的GTP 酶活性丧失,引起腺苷酸环化酶异常激活,继而导致细胞内第二信号cAMP 的过表达,最终引起间充质细胞的前体细胞异常增殖[6-7],导致FD 和IM。由于GNAS1 基因突变发生在体细胞,因此病变组织中突变细胞和正常细胞呈镶嵌分布,而其突变发生的时段和突变细胞的位置、比例决定了发病年龄、病变的范围和严重程度。这从分子机制方面诠释了Mazabraud 综合征临床表现的异质性。目前FD 和IM 的GNAS1基因突变研究多为非Mazabraud 综合征病例,而Mazabraud 综合征极为罕见,迄今对其GNAS1 基因突变研究的文献报道十分有限。国内唐娟等[8]曾研究了1 例Mazabraud 综合征的GNAS1 基因突变情况,在其FD 中发现了该基因的热点突变,即第8 外显子中的第201 位密码子突变为CGT →TGT,但在同一患者的IM 中未检测到突变,究其原因,可能因基因突变细胞太少而导致假阴性结果。Cox 等[9]对其报道的2 例Mazabraud 综合征的其中1 例进行了GNAS1 基因突变分析,发现FD 和IM 均发生了相同的热点突变,即在第201 位密码子均发生了R201H突变。欧洲一项多中心研究对32 例Mazabraud 综合征的流行病学及临床特点进行了回顾性分析,其中在6 例行GNAS1 基因突变检测的IM 中,发现5 例均存在第8 外显子的热点突变,但在这些患者的FD 病变中,因无标本可获取而未能确定FD 病变的突变是否与IM 的突变相匹配[10]。最近,Kašpar 等[11]对其报道的1 例Mazabraud 综合征进行了GNAS1 基因突变分析,在患者的FD 和IM 组织中均检测到了R201H热点突变。这些研究结果进一步证实了GNAS1 基因突变是Mazabraud 综合征的重要病因机制。
Mazabraud 综合征好发于女性,男∶女为1∶2。FD 中位发病年龄为40 岁。根据Vescini 等[12]对Mazabraud 综合征的回顾性研究和文献复习,100 例骨病变中,77 例为多骨性损伤(占77%),常见于股骨和盆骨,也可发生在胫骨、肋骨、颅骨等部位;在单侧和单骨性损伤中,右侧较左侧更多见(57% vs 43%)。IM 发病中位年龄为47 岁,平均比FD 晚6.5年发病,也可单发或多发, 以多发为主(占60%),右侧较左侧更多见(61% vs 39%)。最常见于四肢、肩和臀部肌肉内,尤其大腿最多见,占一半以上。少数情况下,IM 还可见发生在头颈部、胸壁、腹膜后、皮下及关节附近[13]。Hagelstein-Rotman 等[14]报道了来自荷兰、美国和法国三级转诊中心就诊的30 例Mazabraud 综合征患者,发病中位年龄为42 岁,女性20 例(67%),男性10 例(33%),其中26 例患者被诊断为多骨性疾病(87%),29 例患者的IM 位于骨性FD 病变附近(97%)。赵红叶等[15]于2008 年报道了国内首例Mazabraud 综合征,该患者为女性,60 岁,以右大腿腘窝肿块为首发症状,临床表现为右侧股二头肌内近腘窝处单发IM 及同侧股骨上段单骨性FD。其后,国内先后报道了数例Mazabraud 综合征,均为个案报道,年龄从23 岁至70 岁不等,无明显性别差异,全部病例均以软组织肿块为首发症状,多数病例IM 为单发,骨病变中所有病例均累及股骨,约一半病例为多发性骨损伤,左右侧比例基本相同[16-18]。上述临床特点与文献报道有出入,可能与国内病例报道太少而造成的偏差有关。我们报道的这例为57 岁女性患者,FD 表现为多骨型,病变累及左侧股骨上段、髋臼、坐骨及髂骨,即骨性病变见于左侧股骨和左侧盆骨;IM 发生在大腿根部,也是左侧,贴近坐骨结节,临床误认为是坐骨结节囊肿。值得一提的是,解剖学上,本例IM 位于皮下脂肪深层,术中见肿瘤一侧紧贴深筋膜(肌筋膜),而并非发生在肌肉内。根据发现左臀部肿物3 年余的病史,推测IM 发病至少在4 年前;而FD 临床主要表现为近期骨疼,由于之前无手术史、骨折史且无任何骨骼影像学检查,因此无法推测FD 发生的大概年龄及是否早于IM 发病。
Mazabraud 综合征中的FD 其临床症状与病变部位和骨损范围、程度有关,可表现为局部膨隆和肿块、疼痛、畸形等。影像学上,通常表现为界限清楚的低密度膨胀性溶骨性病变,病灶周围骨皮质厚薄不一,边缘有致密硬化,无骨膜反应[19]。FD 典型X 线表现为毛玻璃样外观,周围有致密的骨质,边界清晰,皮质通常完整,但由于膨胀性病变而可能变薄;如果存在软骨岛,在X 线上可呈现爆米花样钙化[20]。MRI 在T1WI 上表现为均匀的低信号,而在T2WI 加权像上表现为混杂信号或高信号,在T1 和T2 加权序列上硬化缘表现为低信号带[21]。CT 主要表现为骨性膨胀伴特征性毛玻璃样改变,有助于更好地显示病变范围,在评价基质矿化、假分隔、扩张、皮质变薄和病理性骨折方面更敏感[22]。ECT 表现为病灶处代谢异常活跃[23]。本病例X 线显示左侧髂骨、左侧股骨上段髓腔内溶骨性病变,密度不均匀,见斑片状低密度影及稍高密度影,呈皂泡样改变,边缘硬化不明显,无骨膜反应。在MRI 上,病变表现为T1WI 低信号混杂点状高信号,T2WI 及压脂序列混杂高信号,DWI 高信号,增强扫描可见不均匀强化。组织学上,正常骨小梁及骨髓被大量增生的纤维样组织所替代,其内散在分布着大小不等、形态不一的骨小梁,排列紊乱、无序,缺乏彼此连接,表面未见明显骨母细胞衬覆。
Mazabraud 综合征中的IM 临床上可无任何症状,通常患者因肿块不断长大而就医,偶尔可伴有疼痛。影像学上,IM 往往发生在FD 区域附近,通常表现为软组织内界限清楚、均质的低密度团块影。X 线呈非特异性软组织肿块,但由于敏感度较低,约一半病例可显示正常结果[24]。MRI 表现包括T1WI上的低信号,T2WI 上的高信号,以及对比增强期的不同模式,包括不均匀或斑片状和周边增强等[25]。CT 通常显示为边界清楚的均匀软组织肿块,其密度高于水,低于周围肌肉组织,大约50%的病例可见轻度弥漫性增强或周边和间隔增强[26]。IM 的这些影像学特征与囊肿或其他黏液样软组织的良性/恶性肿瘤高度相似,鉴别难度较大,但如果存在多个病灶,应该考虑到一种综合征的可能,如Mazabraud 综合征或神经纤维瘤病。我们这例IM 为单发病灶,MRI 表现与文献报道相符,T1WI 为低信号,T2WI 为高信号,增强扫描为环形强化,结合多发性骨损害,影像学上考虑为软组织恶性病变。组织学上,肿瘤组织细胞稀少,呈梭形或星芒状,无异型性,无核分裂像,间质血管稀疏,富含大量黏液样基质;肿瘤与周围组织分界不清,形成不完整的纤维包膜。免疫表型呈vimentin 阳性,CD34、SMA 和Desmin 不同程度表达,S100 阴性[13]。本例除vimentin 阳性外,其余标记均为阴性。IM 属于良性间叶源性肿瘤,基本不恶变,与其他富含黏液、更具侵袭性的病变(如血管黏液瘤、黏液性纤维肉瘤、黏液性脂肪肉瘤等)的鉴别依赖于病变内缺乏高细胞密度、核分裂像和血管。对于形态学及免疫表型不典型的病例,GNAS1基因突变分析将有助于IM的精确诊断[4,27-28]。另外,在熟知Mazabraud 综合征的情况下,相关的FD 的存在也提示软组织病变为良性性质的可能。
Mazabraud 综合征偶可同时伴发McCune-Albright 综合征,后者临床表现为多骨性FD、皮肤咖啡牛奶色素斑和内分泌功能异常[29]。McCune-Albright综合征中的FD 均为多骨性病变,病因也与GNAS1基因突变有关[30]。皮肤色素斑一般分布在有骨病变的同侧。内分泌功能异常可表现为性早熟、甲状腺功能亢进、甲状旁腺功能亢进、生长激素分泌过多、糖尿病、库欣综合征、多囊卵巢和乳腺纤维腺瘤病等。McCune-Albright 综合征发病性别差异与Mazabraud 综合征相似,也好发于女性,女∶男约为2∶1。但根据Biazzo 等[31]的病例报道和文献复习以及后继其他学者的个案报道[32-33],Mazabraud 综合征同时伴发McCune-Albright 综合征的患者几乎均见于女性,13 例中仅1 例为男性,女∶男 > 10∶1,提示两种综合征伴同的患者比任一综合征单发的患者,更常见于女性,但这种性别差异有待更多病例积累证实。
Mazabraud 综合征的治疗方面,对于IM 可行局部手术完整切除,在充分认识这一综合征的情况下,可避免因误诊为恶性而导致的过度扩大切除。FD若为无症状病变,一般不需要治疗,只需连续随访。症状性FD 的治疗通常是非手术治疗,包括双膦酸盐和非甾体抗炎药等用于缓解疼痛[34]。 对于高应力部位(如股骨近端)有症状的、较大的或进行性的病变,以及非手术治疗无效的患者,多主张手术治疗。 FD 手术的目的是预防或治疗畸形和稳定病理性骨折[35]。最常用的手术方法为刮除和植骨术,加或不加内固定[36-37]。但由于手术常难以取得满意效果,目前还没有有效的、成熟的医学治疗方法。最近,有研究显示地舒单抗(denosumab)可直接靶向作用于FD 病变中异位的破骨细胞,促进基质细胞向成骨细胞分化,从而抑制病变发展[38],同时能够有效缓解双膦酸盐治疗无效的FD 患者的疼痛[39],因而denosumab 被认为是治疗FD 前景看好的药物。但停药后导致的反弹相关骨折、颌骨坏死及危及生命的高钙血症等副作用,使denosumab 的临床应用受到限制,后续需要进一步的研究来确定治疗的最佳剂量和持续时间[40]。FD 因经放疗后易诱发恶变,因而禁忌放疗。
Mazabraud 综合征的IM 一般不恶变,但在手术切除后可复发,通常认为其复发率低。但Majoor等[10]对Mazabraud 综合征的20 例IM 患者在手术切除后进行了长期随访,发现其中6 例(30%)复发,复发时间为手术后1.9~16.0 年,中位年限为8.5年。这些研究结果提示Mazabraud 综合征的IM 具有较高的复发率,而且术后复发似乎是一个非常缓慢而持久的过程。孤立性IM 和Mazabraud 综合征的IM 均存在GNAS1 基因突变,提示两者可能具有相同的发病机制。然而,Silver 等[41]对17 例孤立性IM 经手术切除后的患者进行平均7 年(1 至20 年不等)的随访,发现无1 例复发;而Mazabraud 综合征的IM 如前所述,具有相对较高的复发率。这表明两个实体之间存在差异。Mazabraud 综合征中IM的较高复发率可能与组织学特征(如高细胞性)以及多发性IM 以不同速度发展有关,但确切因素仍有待进一步研究。
Mazabraud 综合征的FD 为良性病变,偶可发生恶性转化,形成骨肉瘤、软骨肉瘤或高级别梭形细胞肉瘤[42],但依据报道的整体数据,目前认为其恶变率较高,可达6%[43-44]。因此,对Mazabraud 综合征患者进行影像学长期监测是十分必要的,其中全身MRI 是目前首肯的最佳监测方法。本例经每半年一次的MRI 复查监测,至今随访1 年10 个月,未见IM 复发或FD 恶性转化。
总之,Mazabraud 综合征虽罕见,但影像学表现具有一定特征,最终确诊依赖于病理学检查。与孤立性FD 多发生在儿童和青少年期不同,Mazabraud综合征中的FD 多出现在较晚的成年期。因此,对于发病年龄上可疑的FD 患者,应注意有无合并IM。反之,对于IM 的患者,也须警惕有无FD 存在。遵循从“怀疑”再到“验证”这一诊断思维模式,Mazabraud 综合征诊断的准确性和检出率将会得到进一步提高,从而有效避免误诊、漏诊。
作者声明:本文全部作者对于研究和撰写的论文出现的不端行为承担相应责任;并承诺论文中涉及的原始图片、数据资料等已按照有关规定保存,可接受核查。
学术不端:本文在初审、返修及出版前均通过中国知网(CNKI)科技期刊学术不端文献检测系统的学术不端检测。
同行评议:经同行专家双盲外审,达到刊发要求。
利益冲突:所有作者均声明不存在利益冲突。
文章版权:本文出版前已与全体作者签署了论文授权书等协议。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
中华骨与关节外科杂志(2021年12期)2021-08-31 09:10:54
中国生殖健康(2020年2期)2021-01-18 02:51:26
中国临床医学影像杂志(2019年5期)2019-08-27 02:48:00
中国临床医学影像杂志(2019年2期)2019-04-25 06:15:50
小学生导刊(2018年13期)2018-06-29 03:49:00
现代检验医学杂志(2016年3期)2016-11-15 01:59:26
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:56
中国继续医学教育(2015年3期)2016-01-06 01:36:36
分子影像学杂志(2015年3期)2015-12-04 03:28:58
