
锈赤扁谷盗的全长转录组测序分析
2023-10-04 18:32胡怀月陈二虎韦磊王康旭李孟凡唐培安
粮食科技与经济 2023年3期
胡怀月 陈二虎 韦磊 王康旭 李孟凡 唐培安
摘要:锈赤扁谷盗是一种世界性储粮害虫,但对其分子生物学方面的基础研究较少。研究基于PacBio IsoSeq平台对锈赤扁谷盗进行全长转录组测序,并对其数据进行生物学分析。测序数据经过校正后,锈赤扁谷盗全长转录组平均长度为1 845.06 bp,N50长度为2 130 bp;经去冗余后,获得转录本32 855条。在Nr、KEGG、KOG、Swiss-Prot四大数据库中,分别有29 404、27 794、20 286、21 197条转录本被注释;其中,19 346条转录本在4个数据库中均有注释,29 416条转录本至少在一个数据库中有注释,占总数的89.53%。此外,经鉴定,获得1 139个转录因子(TFs)、2 636条长链非编码RNA(LncRNA)和4 197个SSR,预测后获得32 800条CDS。
关键词:锈赤扁谷盗;全长转录组;基因组注释
中图分类号:S379.5 文献标志码:A DOI:10.16465/j.gste.cn431252ts.20230326
基金项目:国家重点研发计划专项(2021YFD2100604-01);江苏省重点研发计划项目(BE2022377);国家自然科学基金项目(32272388);江苏省高校优势学科建设工程资助(PAPD);江苏省研究生科研与实践创新计划(KYCX21_1528)。
Sequencing analysis of the full-length transcriptome data of Cryptolestes ferrugineus
Hu Huaiyue1, Chen Erhu1, Wei Lei2, Wang Kangxu1, Li Mengfan1, Tang Peian1
( 1. College of Food Science and Engineering, Nanjing University of Finance and Economics/ Collaborative Innovation Center for Modern Grain Circulation and Safety, Nanjing, Jiangsu 210023; 2. Jiangsu Wanjiafu Rice Industry Co., Taizhou, Jiangsu 225700 )
Abstract: Cryptolestes ferrugineus is a worldwide grain storage pest, but there are few basic studies on its molecular biology. In this study, we sequenced the full-length transcriptome of C. ferrugineus based on PacBio Iso-Seq platform and analyzed the data biologically. After the sequencing data were corrected, the average length of the full-length transcriptome was 1 845.06 bp, and the N50 length was 2 130 bp; after redundancy removal, 32 855 transcripts were obtained. Among the four major databases, Nr, KEGG, KOG and Swiss-Prot, 29 404, 27 794, 20 286 and 21 197 transcripts were annotated respectively; among them, 19 346 transcripts were annotated in all four databases, and 29 416 transcripts were annotated in at least one database, accounting for 89.53% of the total. In addition, 1 139 transcription factors (TFs), 2 636 long-stranded non-coding RNAs(LncRNAs) and 4 197 SSR were identified, and 32 800 CDS were obtained after prediction.
Key words: Cryptolestes ferrugineus, full-length transcriptome, genome annotation
銹赤扁谷盗Cryptolestes ferrugineus(Stephens)属于鞘翅目扁谷盗科,主要分布在温带和热带地区。在全球范围内,锈赤扁谷盗是发生最严重、最广泛的储粮害虫之一,一旦发生便会造成严重的不可逆损失[1-2]。锈赤扁谷盗在我国主要分布于粮食生态区,在广东、云南和海南等地都有发生,常见于米厂、面粉厂、酒厂和一些粮仓内[3]。锈赤扁谷盗在粮堆的分布也受温度和湿度的影响[4],在粮堆发热的情况下,更容易造成锈赤扁谷盗的繁殖,严重时可出现粮堆结露,进而使粮食发生霉变,给储粮安全带来极大的威胁[5]。
尽管二代测序高通量测序技术对转录组学的相关研究起到了极大的推动作用,但还是存在测序读长较短[6]、重复区域拼接效果差、无法得到较完整的转录本[7]等局限性。近年来,与第二代测序相比,三代转录组测序可快速全面地获得某一物种的全长转录本信息,逐渐被应用于昆虫学研究中,例如,按蚊[8]、家蚕[9]、小菜蛾[10]等,主要集中在生长发育抗药性和遗传变异等方面。在锈赤扁谷盗的研究中,关于三代测序的报道相对较少,因此,本文对锈赤扁谷盗进行全长转录组测序并通过基因功能注释、CDS预测、TFs分析等,进一步从分子水平对锈赤扁谷盗的研究奠定基础,也为探究新的靶标基因、利用分子设计实现害虫防治提供理论依据并丰富鞘翅目昆虫的全长转录组数据库。
1 材料与方法
1.1 供试昆虫
本试验锈赤扁谷盗种群采自上海福星面粉厂,于南京财经大学粮食储运国家工程实验室培养。在实验室内以人工饲料(全麦粉∶燕麦∶小麦碎∶酵母粉=5∶3∶3∶1)、恒温29~31 ℃、相对湿度为70%~80% RH、无光照条件下饲养。在培养过程中,锈赤扁谷盗以3 d产的卵为同一龄期,每隔3 d将瓶中锈赤扁谷盗成虫全部挑出放入相同饲料的新瓶子中,重复此操作4~5次之后即可获得龄期一致的试虫。
1.2 方法
1.2.1 总RNA提取及文库构建
对锈赤扁谷盗幼虫、蛹、成虫3个阶段分别进行取样,每个阶段3个重复,对混合样品进行RNA提取和富集,使用NanoDrop分光光度计检测样品RNA的浓度;使用Agilent2100检测样品RNA的完整度。总RNA(含有poly(A))检测合格后,利用Clontech SMARTer PCR cDNA Synthesis Kit反转录成第一链cDNA,PCR扩增合成双链cDNA并对PCR产物进行纯化,对cDNA进行DNA损伤修复、末端修复、连接Adapter和文库质量评估工作后形成完整的SMRT bell文库,再对文库进行测序。
1.2.2 全长转录组数据分析和校正
使用SMRT Link V8.0原始数据进行分析。首先从下机数据中提取高质量的CCS(circular consensus sequence)序列,移除引物和barcode、poly(A)和连环结构,得到全长非嵌合序列(FLNC),再对相似的FLNC reads进行聚类,合并成一个完整的转录本序列。使用LoRDEC软件进行转录本校正,LoRDEC采用混合策略,需要使用两组数据:参考reads(二代短reads)、PacBio长reads。通过读取短reads,建立DBG图,针对长reads中的错误区域寻找校正序列。之后,通过Nr、KEGG、KOG、Swiss-Prot、Pfam和GO数据库分别对锈赤扁谷盗全长转录组进行功能注释,并进行转录因子(TFs)鉴定、长链非编码RNA(lncRNAs)预测和SSR分析等。
2 结果与分析
2.1 测序数据分析
测序结果表明,锈赤扁谷盗全长转录组数据库包含414 552条CCSs,平均长度为1 868 bp。此外,结果显示含有12 009 936条子序列,子序列平均长度为1 845 bp,N50长度为2 094 bp。在聚类和校正之后,对序列进行去冗余,获得32 855条转录本,平均长度为1 845.06 bp(如图1),N50长度为2 130 bp,如表1。
2.2 功能注释
2.2.1 基本注释
对得到的32 855条转录本进行Nr、KEGG、KOG、Swiss-Prot四大数据库注释,如图2所示,其中Nr数据库中有29 404条转录本被注释,KEGG数据库中有27 794条转录本被注释,KOG数据库中有20 286条转录本被注释,SwissProt数据库中有21 197条转录本被注释,其中至少被一个数据库注释的转录本数目为29 416条(89.53%),19 346条转录本在四大数据库中都有注释。
2.2.2 E值分布
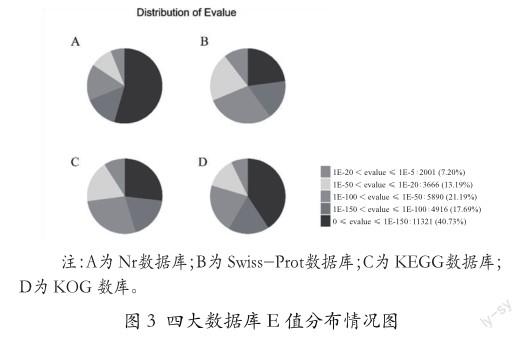
将所有转录本在 Nr、Swiss-Prot、KEGG 和KOG 四大数据库中的最佳比对结果的E值进行统计,将其分为5个范围。由E值可以看出,转录与数据库中匹配序列为同源序列的假阳性概率。如图3所示,Nr数据库中,在0≤E值≤1E-150范围内有16 004条转录本,占54.43%;SwissProt数据库中,在0≤E值≤1E-150范围内有4 843条转录本,占22.85%;KEGG数据库中,在0≤E值≤1E-150范围内有11 321条转录本,占40.73%;KOG 数库中,在0≤E值≤1E-150范围内有5 454条转录本,占26.89%。
2.2.3 物種分布
利用Blastx将转录本序列与Nr数据库进行比对后,取每个转录本在Nr数据库中比对结果最好(E值最低)的那一条序列作为对应同源序列确定同源序列所属物种,统计比对到各个物种的同源序列数量,同源性较高的前3位分别是光肩星天牛、赤拟谷盗和蜂箱小甲虫,如表2所示。
2.2.4 GO分类
在GO数据库中获得注释转录本173 206条,如图4所示,生物学过程(biological process)包括25个功能组,其中细胞过程(cellular process)包含转录本最多,有13 585条,其次是单一生物过程(single-organism process)12 740条,最少的是生物阶段(biological phase),只有11条;细胞组分(cellular component)包括21个功能组,其中细胞(cell)和细胞组分(cell part)包含转录本最多,都有11 053条,最少的是拟核(nucleoid),只有1条;分子功能(molecular function)包括12个分子功能,其中结合活性(binding)最多,有10 176条,其次是催化活性(catalytic activity)9 217条,电子载体活性(electron carrier activity)最少,只有14条。
2.3 高级注释及基因结构分析
2.3.1 TFs鉴定
将预测的蛋白序列同相应的TF数据库(Animal TFdb)进行 hmmscan 比对。结果显示,一共预测到1 139个转录因子,属于55个TF家族,对转录本数目最多的前10个TF家族进行绘图,如图5所示。其中Zf-C2H2家族有355个、ZBTB家族有74个、TF_bzip家族有72个,HMG家族有71个。
2.3.2 LncRNA预测
使用CNCI软件和CPC软件进行编码能力预测,取两个软件都预测为“非编码”的结果作为最终的lncRNA结果。如图6所示,CNCI预测lncRNA 2 871个,CPC预测lncRNA 2 846个,两个软件同时预测lncRNA 2 636个。
2.3.3 SSR分析
利用MISA软件对转录组的所有转录本进行搜索,寻找转录本中的SSR,分析结果显示,不同串联重复单元类型的SSR在总SSR中所占比例不同,其中AAT/ATT的频率最高,所占比例为31.3%。如图7所示,在SSR核苷酸基序类型中,三核苷酸重复最多(tri nucleotides,69.19%),依次是四核苷酸重复(tetra nucleotides,5.23%)、双核苷酸重复(di nucleotide motifs,13.27%)、五核苷酸重复(penta nucleotides,2.14%)和六核苷酸重复(hexa-nucleotide motifs,0.17%)。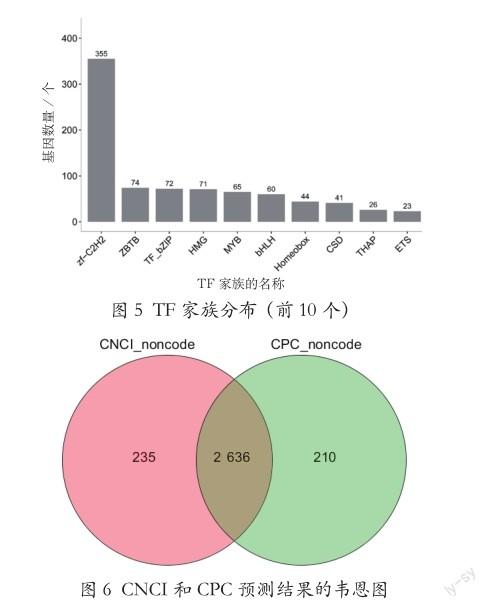
3 讨论与结论
二代测序虽在转录组学相关研究中应用广泛,但存在测序读长短,对高重复区域无法较好地拼接等缺点。相比于二代转录组测序定量的特点,全长转录组还可对转录本进行定性分析,即分析转录本的结构。第三代测序技术具有超长读长、无需组装即可直接获得RNA的全长序列,还可鉴定基因的可变剪切、可变多聚核苷酸化APA、非编码RNA等,让测序的转录组数据更加丰富可靠。
本研究利用第三代测序技术对锈赤扁谷盗不同龄期混合样品进行测序,校正后去冗余共获得32 855条转录本,平均长度为1 845.06 bp,N50长度为2 130 bp。通过在Nr、KEGG、KOG、Swiss-Prot四大数据库中注释,至少被一个数据库注释的转录本数目为29 416条,占总转录本的89.53%,被注释到Nr数据库的最多,有29 404条。而对莲草直胸跳甲卵、幼虫、蛹、成虫4个发育阶段的样本的全长转录组测序,共获得28 982条高质量的转录本,并预测得到4 198个lncRNA,24 040个开放阅读框ORF[11];对家蚕丝腺的测序获得11 697条转录本[12]。lncRNA具有调控转录、剪切或相邻基因的表达等功能。例如在果蝇中,lncRNA可以调控雄性果蝇的性别决定[13]、睡眠[14]、运动行为[15]等生物过程;在家蚕丝腺中发现了一些与蚕丝基因表达相关的lncRNA[16]。近年来,大量的lncRNA已从家蚕[16]、黑腹果蝇[17]、小菜蛾、冈比亚按蚊、中华蜜蜂[18]等昆虫中鉴定出来。本文共获得2 636个lncRNA,为后续在基因表達和调控提供参考。在锈赤扁谷盗中共获得1 139个转录因子,Zf-C2H2家族有355个、ZBTB家族有74个、TF_bzip家族有72个,HMG家族有71个。不同的TFs可能参与不同的代谢过程,也可能有多种不同的功能[19]。在很多研究中发现,转录因子与解毒酶基因的表达相关。例如,在赤拟谷盗中,转录因子CncC和Maf通过调控CYP6BQ基因上调增强其对溴氰菊酯的代谢[20];转录因子FTZ-F1可以调控小菜蛾CYP6BG1基因的表达,从而使小菜蛾对氯虫苯酰胺的代谢增强[21]。所以,对锈赤扁谷盗的TFs分析会对其免疫和抗药性方面的分析提供依据。通过对SSR开发标记发掘其功能,也为锈赤扁谷盗遗传多样性分析提供更多数据支持。
综上,本文基于全长转录组测序技术获得了锈赤扁谷盗的全长转录组,并注释了锈赤扁谷盗全长转录组相关基因功能信息,也为进一步研究锈赤扁谷盗的防治筛选靶标,并为其遗传机制的研究奠定基础。
参 考 文 献
[1]PHILLIPS T W, THRONE J E. Biorational approaches to managing stored-product insects[J]. Annual review of entomology, 2010, 55: 375-397.
[2] LOSEY S M, DAGLISH G J, PHILLIPS T W. Orientation of rusty grain beetles, Cryptolestes ferrugineus (Coleoptera:Laemophloeidae), to semiochemicals in field and laboratory experiments[J]. Journal of Stored Products Research, 2019, 84: 101513.
[3] 张瑞杰,王殿轩,白春启,等.中国12省80地市储粮中扁谷盗属昆虫分布调查[J].河南工业大学学报(自然科学版),2018, 39(5):112-118.
[4] JIAN F J, LARSON R, JAYAS D S, et al. Three-dimensional spatial distribution of adults of Cryptolestes ferrugineus (Coleoptera: Laemophloeidae) in stored wheat under different temperatures, moisture contents, and adult densities[J]. Journal of Stored Products Research, 2011, 47(4): 293-305.
[5] PARDE S R, JAYAS D S, WHITE N D G. Movement of Cryptolestes ferrugineus (Coleoptera: Cucujidae) in grain columns containing pockets of high moisture content wheat and carbon dioxide gradients[J]. Journal of Stored Products Research, 2004, 40(3): 299-316.
[6] KOREN S, SCHATZ M C, WALENZ B P, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads[J]. Nature Biotechnology, 2012, 30(7): 693-700.
[7]田李,張颖,赵云峰.新一代测序技术的发展和应用[J].生物技术通报,2015,31(11):1-8.
[8] JIANG X, HALL A B, BIEDLER J K, et al. Single molecule RNA sequencing uncovers trans‐splicing and improves annotations in Anopheles stephensi[J]. Insect Molecular Biology, 2017, 26(3): 298-307.
[9] KAWAMOTO M, JOURAKU A, TOYODA A, et al. High-quality genome assembly of the silkworm, Bombyx mori [J]. Insect Biochemistry and Molecular Biology, 2019, 107: 53-62.
[10] ZHAO Q, ZHONG W M, HE W Y, et al. Genome-wide profiling of the alternative splicing provides insights into development in Plutella xylostella [J]. BMC Genomics, 2019, 20(1): 1-14.
[11] JIA D, WANG Y X, LIU Y H, et al. SMRT sequencing of fulllength transcriptome of flea beetle Agasicles hygrophila (Selman and Vogt)[J]. Scientific Reports, 2018, 8(1): 1-8.
[12] CHEN T, SUN Q W, MA Y, et al. A transcriptome atlas of silkworm silk glands revealed by PacBio single-molecule long-read sequencing[J]. Molecular Genetics and Genomics, 2020, 295(5): 1227-1237.
[13] MULVEY B B, OLCESE U, CABRERA J R, et al. An interactive network of long non-coding RNAs facilitates the Drosophila sex determination decision[J]. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms, 2014, 1839(9): 773-784.
[14] SOSHNEV A A, ISHIMOTO H, MCALLISTER B F, et al. A conserved long noncoding RNA affects sleep behavior in Drosophila[J]. Genetics, 2011, 189(2): 455-468.
[15] LI M X, WEN S Y, GUO X Q, et al. The novel long non-coding RNA CRG regulates Drosophila locomotor behavior[J]. Nucleic Acids Research, 2012, 40(22): 11714-11727.
[16] WU Y Q, CHENG T C, LIU C, et al. Systematic identification and characterization of long non-coding RNAs in the silkworm, Bombyx mori[J]. PloS one, 2016, 11(1): e0147147.
[17] YOUNG R S, MARQUES A C, TIBBIT C, et al. Identification and properties of 1,119 candidate lincRNA loci in the Drosophila melanogaster genome[J]. Genome Biology and Evolution, 2012, 4(4): 427-442.
[18] JAYAKODI M, JUNG J W, PARK D, et al. Genome-wide characterization of long intergenic non-coding RNAs (lincRNAs) provides new insight into viral diseases in honey bees Apis cerana and Apis mellifera[J]. BMC Genomics, 2015, 16(1): 1-12.
[19] CHEN K, RAJEWSKY N. The evolution of gene regulation by transcription factors and microRNAs[J]. Nature Reviews Genetics, 2007, 8(2): 93-103.
[20] KALSI M, PALLI S R. Transcription factors, CncC and Maf, regulate expression of CYP6BQ genes responsible for deltamethrin resistance in Tribolium castaneum[J]. Insect Biochemistry and Molecular Biology, 2015, 65: 47-56.
[21] LI X X, SHAN C Y, LI F, et al. Transcription factor FTZ‐F1 and cis‐acting elements mediate expression of CYP6BG1 conferring resistance to chlorantraniliprole in Plutella xylostella[J]. Pest Management Science, 2019, 75(4): 1172-1180.
